
Research
Read about the Wilson Lab’s core areas of research:
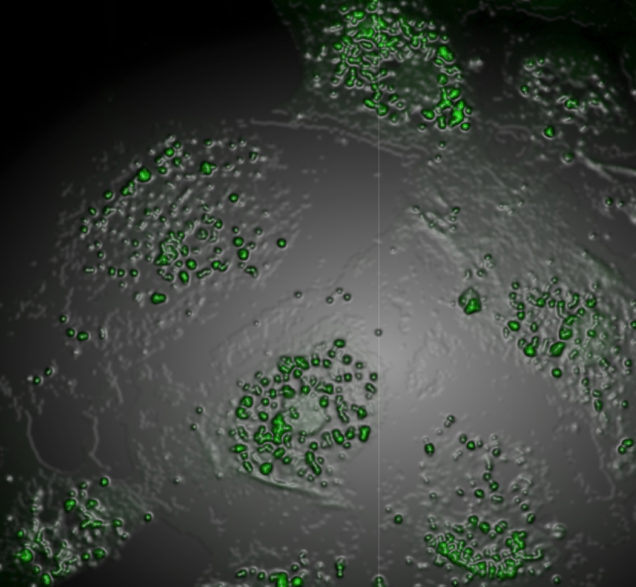
Confirm the clinical significance of the iPSC platform to model in vivo patient biology and demonstrate its potential for testing potential therapeutic agents:
iPSCs were first described in 2006 and have been used to model a variety of diseases. We and others have demonstrated the ability of AATD patient-iPSCs to recapitulate salient AATD disease features in vitro. It is widely believed that patient-iPSCs and their differentiated progeny can act as surrogates for the patients from whom they were derived, a finding that, if true, would make possible personalized predictions of drug efficacy and toxicity. The Wilson Lab is testing the hypothesis that drug responsiveness of iPSC-derived cell types such as hepatocytes predict the response of an individual patient’s hepatocytes in vivo.
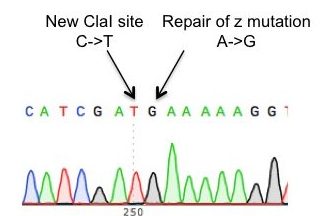 Understand the genetic factors and mechanistic drivers that predispose subsets of AATD patients to develop clinical disease:
Understand the genetic factors and mechanistic drivers that predispose subsets of AATD patients to develop clinical disease:
It is well described that only ~15% of individuals homozygous for the single base pair “Z” mutation (homozygotes are referred to as “PiZZ”) go on to develop clinically significant liver disease and that genetic cofactors likely contribute to disease susceptibility. Similarly, while PiZZ patients are highly predisposed to developing emphysema, significant heterogeneity exists in the degree of damage observed, with a subset of patients retaining normal lung function throughout their lives. We and others have demonstrated that the differentiated progeny of patient-derived iPSCs model key features of AATD-associated cellular disease. To define a disease signature based on these cells, the Wilson Lab has developed an approach using the CRISPR/Cas9 nuclease to correct the “Z” mutation in patient-derived iPSCs, allowing us to compare isogenic lines derived from a single individual that differ only at the SERPINA1
gene that encodes AAT. We are now exploring the potential contribution of genes from this signature to disease susceptibility and evaluating differences in the disease signature produced by PiZZ vs gene corrected iPSCs from individuals with distinct clinical phenotypes.
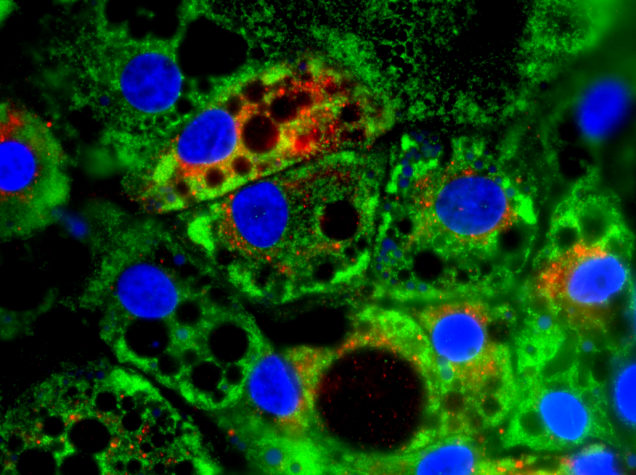
Elucidate the mechanistic contribution of putative COPD susceptibility genes to lung disease pathogenesis:
A limited understanding of COPD pathophysiology has hampered the efforts of researchers to develop new potential therapies. COPD develops only in a small percentage of long-term smokers, clusters in families, and exhibits variable prevalence in different racial populations, suggesting a significant contribution of genetic factors to disease susceptibility. In the past two decades, an increasing number of genes have been identified by GWAS studies as potential contributors to the disease process but their mechanistic contributions to disease remain unknown. To address this challenge, the Wilson Lab is combining patient iPSCs and CRISPR-based gene manipulation to test the mechanistic contribution of genes associated with COPD risk in GWAS studies to lung development or response to injury.
Develop gene or cell-based therapies for AATD:
AATD patients have low levels of circulating AAT protein, predisposing them to early onset emphysema from an imbalance in protease and antiprotease 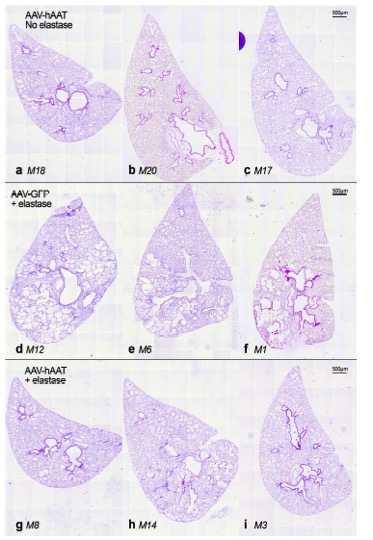 activity in the lungs. To combat lung damage, patients with AATD are treated with lifelong infusions of pooled human plasma AAT. The invasive nature of this treatment, its expense, and its requirement for lifelong exposure to human blood products support the need for alternative therapies. Supplying patients with normal AAT gene expression and protein production, either through vector-based gene delivery or delivery of engraftable cells expressing the normal AAT gene is an approach that could circumvent the shortcomings of current treatment approaches. The Wilson Lab has demonstrated the ability of AAT gene therapy to protect animals against experimental emphysema and determined that lung-localized transgene delivery is more efficient than conventional gene delivery to peripheral tissues.
activity in the lungs. To combat lung damage, patients with AATD are treated with lifelong infusions of pooled human plasma AAT. The invasive nature of this treatment, its expense, and its requirement for lifelong exposure to human blood products support the need for alternative therapies. Supplying patients with normal AAT gene expression and protein production, either through vector-based gene delivery or delivery of engraftable cells expressing the normal AAT gene is an approach that could circumvent the shortcomings of current treatment approaches. The Wilson Lab has demonstrated the ability of AAT gene therapy to protect animals against experimental emphysema and determined that lung-localized transgene delivery is more efficient than conventional gene delivery to peripheral tissues.
We have likewise developed methods for footprint free gene editing of patient iPSCs and used them to correct the Z mutation responsible for AATD in patient cells. We are now collaborating with colleagues at UMass Medical School to develop methods for transplant of gene-corrected patient iPSCs and to test their capacity to engraft and function in the livers of recipient animals. If successful, this line of research will represent an important first step towards a cure for patients with AATD.