
Electron Transfer in Organic Radicals
By Nifemi Mabayoje and De’Ja Rogers
Stable organic radicals present a promising solution to future energy needs. Current batteries rely on the use of rare earth metals such as lithium, which have a high cost to energy ratio. Organic radical batteries may provide a cheaper, more sustainable alternative. 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) is a stable organic radical that can be polymerized into poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA) and can be used to create a cathode. This organic radical polymer can participate in redox reactions that can produce an electrochemical potential and be used for energy storage.
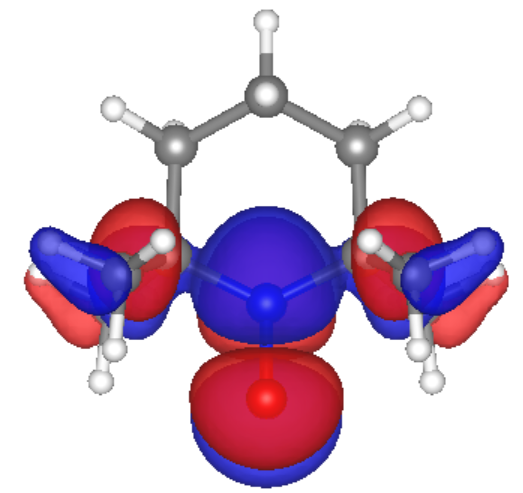
Singularly occupied molecular orbital (SOMO) of TEMPO
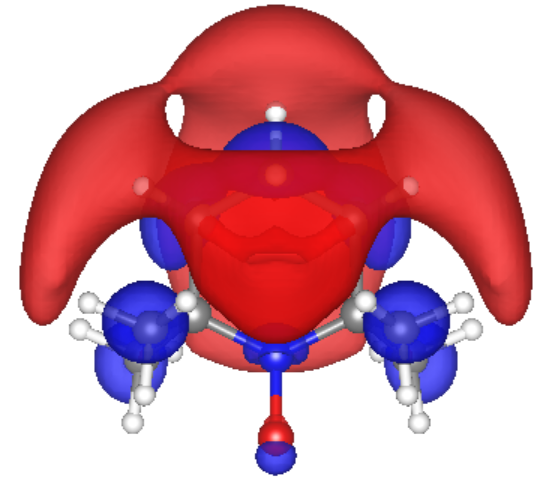
Lowest unoccupied molecular orbital (LUMO) of TEMPO
The purpose of our research is to study the relationship between the structure of PTMA and the rate of electron transfer in the polymer. Density Functional Theory (DFT) and Hartree Fock, both quantum mechanical simulation methods, were used to analyze five different arrangements of TEMPO on PTMA polymers in order to see which polymer structure led to the most efficient electron transfer.
Our first step was to use Avogadro, a molecular modelling software, to create the PTMA structures. We considered five different structures: a monomer (case 0), a dimer case with the TEMPO groups side by side and parallel on the backbone (case 1), a dimer with the TEMPO rings at a 180 degree angle from each other, on opposite sides of the chain (case 2), a dimer with the TEMPO groups at 90 degrees from each other (case 3), a dimer with two polymer chain spacings between the TEMPO rings (case 4), and a trimer with two rings on one chain and one TEMPO ring on the other chain, the chains facing each other in a zipper-like configuration. We include a PTMA monomer to understand the behavior of PTMA without the interactions between different TEMPO rings.
Since neither of us was familiar with first principles, quantum mechanical calculations, we also took a course on NanoHub to familiarize ourselves with quantum mechanics and its applications in material design. NanoHub is an educational website containing courses and tools for disseminating knowledge in the field of nanoscience. NanoHub’s simulation tools were also a great place to begin exploring free simulation tools and familiarizing ourselves with examining the data from DFT calculations.
After spending some time on NanoHub, we ran into some limitations as the tools available on NanoHub could not simulate high spin/open shell systems. So we began to learn the Bash programming language and Linux, so that we could move our work to Boston University’s shared computing cluster where we could use Gaussian09 and Gaussview to run simulations and view the results. We used Gaussian as a tool to calculate orbital energies and total energies, and visualize molecular orbital diagrams and charge density for all cases. For each case, we simulated both spin 0 and unrestricted high spin cases. We used Density Functional Theory and Hartree Fock for our simulations; the basis sets used include CAM-B3LYP, B3LYP, and Hartree Fock at 6-31G(d,p).
Our calculations and simulations produced promising results. The molecular orbitals of the monomer case show that the frontier orbitals are mostly located at the TEMPO ring, which would make the electron hopping from one radical site to another feasible. Also, we calculated reorganization energy from simulations for the monomer ion. Our result for the monomer cation reorganization energy, 1 eV, agrees with previous research by Kemper et al.
The spin density of the dimer cases consistently show the radical electrons reside primarily at the N-O bonds. In case 5, the Trimer case, the unpaired electrons are delocalized throughout the PTMA wires. Case 5 also indicates hybridization of the molecular orbitals due to the radical electrons’ unique interactions, while the dimer cases resemble two non-interacting monomers, side by side. In the trimer case, the average rate of electron transfer was calculated using Marcus Theory to be 4 X 109 s-1 and the average electron mobility was calculated to be 2.2 x 10-4 cm2 V-1 s-1. These values agree with the reported experimental values for electron transfer in PTMA polymer.
Of the six cases, we found that case 5, representing inter-chain electron hopping, is the best choice for electron transfer. This is because of the hybridization of the radical electrons in the molecular orbitals, and the delocalization of the electrons in the spin density. Our electron transfer rate and electron mobility values for inter-chain hopping are also corroborated by experimental data. A possible area for further exploration would be working on larger systems and other similar configurations to further understand the relationship between the structure and function of PTMA.