Blog Posts
A DFT Analysis of Raman Signatures of Pt-Au Interactions in Quasi-One Dimensional Metal-Organic Nanowires

Metallophilic interactions between closed shell metal ions with electron configurations, d8, d9 and d10, are widely studied, because these interactions are essential to the behavior of molecular conductors in molecular electronics. Metal-metal interactions occur between atoms with single valence electrons, where the valence and conduction bands of both metals, in this research, Pt+ and Au-, might express an overlap in energetic bands. When closely packed into a crystal structure, atoms’ valence electrons will overlap causing electrons to delocalize forming a free electron system. A conductive material will have an overlap in energy bands and electron delocalization. Therefore, Pt+ and Au-, when close in proximity, have closed-shell interactions that makes them effective as conductors, similar to the weak effects of Van der Waals interactions.
We can understand these interactions using vibrational spectra. Raman spectroscopy uses photons to shift the crystal into a temporary state of excitation. If there is an overlap in energetic bands, meaning metal-metal interactions between the metallic core atoms and their valence electrons, this state of temporary excitement will exhibit a noticeable attraction between the two metallic cores. Therefore, Raman spectroscopy is an effective tool to measure metallophilic interactions.
This research studies the vibrational properties of tunable quasi-one-dimensional metal- organic chains that are promising as electronic and spintronic materials. These structures consist of single chains of the form [Pt(tpy)X][AuY2] that self-assemble as weakly bound wires within an anisotropic crystal in the solid-state, forming salts of alternating Pt+ and Au- atoms embedded in organic matrices [Hayoun, R.; Zhong, D. K.; Rheingold, A. L.; Doerrer, L. H. Inorganic Chem., 2006, 45(16), 6120-6122]. Here, we utilize density functional theory (DFT) and Raman spectroscopy (as implemented in Gaussian 09 and Quantum Espresso) in order to probe their structural and vibrational properties. By comparison of prediction with measurements, we determine the optical signatures of intra-chain, specifically the Au… Pt mode found experimentally at 140cm-1, and inter-chain interactions of the quasi-one dimensional crystals.
We used calculated frequency spectra of 6 different clusters, where results with and without periodic boundary conditions were compared to experiment. Our simulations do not contain the packing forces of infinite neighboring chains which would exist experimentally. Therefore, geometry optimization caused the metallic cores to expand from 3.349Å to 4.067Å, a distance too large for metal-metal contacts to form; thus, we used experimental geometry. The different clusters allowed us to identify vibrational modes of intra-chain, inter-chain and anion-cation interactions within the wires. The single chain clusters allowed us to identify the modes corresponding to intra-chain and anion-cation interactions, while the double chain clusters allowed us to identify inter-chain interactions and effects that neighboring chains might have on the vibrational spectra.
We first calculated the Raman spectra of our single chain two aperiodic-units cluster and double chain two aperiodic-units cluster using Gaussian 09. B3LYP was used to calculate the Raman spectra of the single chain two aperiodic-units cluster. B3LYP with dispersion correction (IOp(3/124=3) was used to calculate the Raman spectra of the double chain two aperiodic-units cluster in order to include Van der Waals forces.
We proceeded to calculate the vibrational spectra of both clusters with periodic boundary conditions. Parameters used in Quantum Espresso were as follows: LDA functional and norm- conserving pseudopotentials in separable (Kleinman-Bylander) form, gamma-only, free structure with specified cell parameters. Atomic coordinates were the same as the experimental coordinates used in the structures of the aperiodic calculations, for consistency. Dispersion correction was included as Grimme-D2 scheme. Generally, total energy convergence can be achieved with a rather low wave function cutoff of 120Ry; however, we found that the calculated spectra varied greatly from 120Ry to 300Ry. Due to computational limitations, we were not able to compute vibrational modes with a wave function cutoff higher than 300Ry. Feasible total energy convergence studies allowed us to choose a suitable wave-function cutoff (300 Ry) and k- point density (9x5x3). Similar to the aperiodic calculations, we did not optimize the geometry and instead opted for experimental geometry.
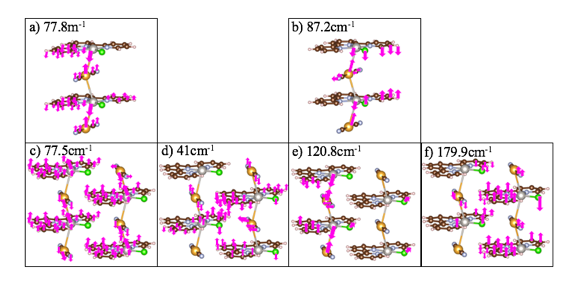
We calculated inter-chain interactions for the double chain two aperiodic-units clusters and double chain two periodic-units cluster at 41cm-1 and 179.9 cm-1 respectively. The cation-anion modes for the single chain two aperiodic-units cluster, single chain two periodic-units cluster, and double chain two aperiodic-units cluster, were found at 77.8 cm-1, 87.2 cm-1 and 77.5 cm-1 respectively. As for the double chain two periodic-units cluster, theoretical results found intra-chain modes for cation-anion interactions in the range, 115-148 cm-1, similar to the experimental peak, 140 cm-1. Furthermore, the terpyridine peaks in the range, 700-1650 cm-1, are blue shifted for the theoretical spectrum compared to the experimental, which is also why we observe cation-anion interactions in a slightly lower range as well. These results are consistent and a promising start to confirming metallophilic interactions and ultimately, conductivity.
In conclusion, the double chain two-periodic units cluster yielded the most accurate results for Raman vibrational modes of the [Pt(tpy)Cl]+…[Au(CN)2]- self-assembled nanowires compared to experiment. This is to be expected, because it is most closely related to experiment environment in terms of neighboring chains and periodicity. The aperiodic calculations and single chain two periodic-units calculations produced cation-anion modes much lower than experiment, 140 cm-1. To produce the most accurate DTF calculations, it is necessary to calculate the spectra of the bulk structure, or even a cluster with 4-plus adjacent chains; for, periodic boundary conditions had a substantial effect on the vibrational spectra.
Electron Transfer in Organic Radical Batteries
By Nifemi Mabayoje and De’Ja Rogers

Stable organic radicals present a promising solution to future energy needs. Current batteries rely on the use of rare earth metals such as lithium, which have a high cost to energy ratio. Organic radical batteries may provide a cheaper, more sustainable alternative. 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) is a stable organic radical that can be polymerized into poly(2,2,6,6-tetramethylpiperidinyloxy methacrylate) (PTMA) and can be used to create a cathode. This organic radical polymer can participate in redox reactions that can produce an electrochemical potential and be used for energy storage.
The purpose of our research is to study the relationship between the structure of PTMA and the rate of electron transfer in the polymer. Density Functional Theory (DFT) and Hartree Fock, both quantum mechanical simulation methods, were used to analyze five different arrangements of TEMPO on PTMA polymers in order to see which polymer structure led to the most efficient electron transfer.
Our first step was to use Avogadro, a molecular modelling software, to create the PTMA structures. We considered five different structures: a monomer (case 0), a dimer case with the TEMPO groups side by side and parallel on the backbone (case 1), a dimer with the TEMPO rings at a 180 degree angle from each other, on opposite sides of the chain (case 2), a dimer with the TEMPO groups at 90 degrees from each other (case 3), a dimer with two polymer chain spacings between the TEMPO rings (case 4), and a trimer with two rings on one chain and one TEMPO ring on the other chain, the chains facing each other in a zipper-like configuration. We include a PTMA monomer to understand the behavior of PTMA without the interactions between different TEMPO rings.
Since neither of us was familiar with first principles, quantum mechanical calculations, we also took a course on NanoHub to familiarize ourselves with quantum mechanics and its applications in material design. NanoHub is an educational website containing courses and tools for disseminating knowledge in the field of nanoscience. NanoHub’s simulation tools were also a great place to begin exploring free simulation tools and familiarizing ourselves with examining the data from DFT calculations.
After spending some time on NanoHub, we ran into some limitations as the tools available on NanoHub could not simulate high spin/open shell systems. So we began to learn the Bash programming language and Linux, so that we could move our work to Boston University’s shared computing cluster where we could use Gaussian09 and Gaussview to run simulations and view the results. We used Gaussian as a tool to calculate orbital energies and total energies, and visualize molecular orbital diagrams and charge density for all cases. For each case, we simulated both spin 0 and unrestricted high spin cases. We used Density Functional Theory and Hartree Fock for our simulations; the basis sets used include CAM-B3LYP, B3LYP, and Hartree Fock at 6-31G(d,p).
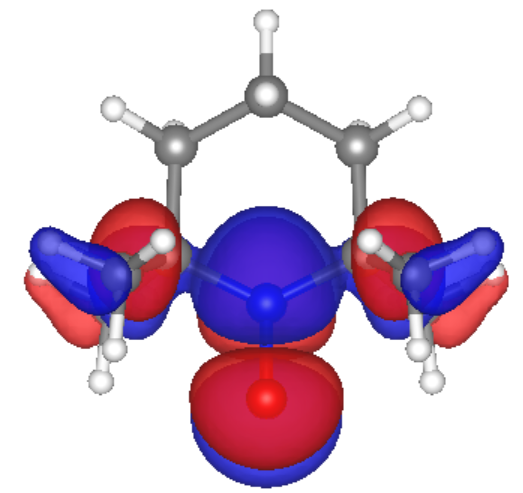
Our calculations and simulations produced promising results. The molecular orbitals of the monomer case show that the frontier orbitals are mostly located at the TEMPO ring, which would make the electron hopping from one radical site to another feasible. Also, we calculated reorganization energy from simulations for the monomer ion. Our result for the monomer cation reorganization energy, 1 eV, agrees with previous research by Kemper et al.
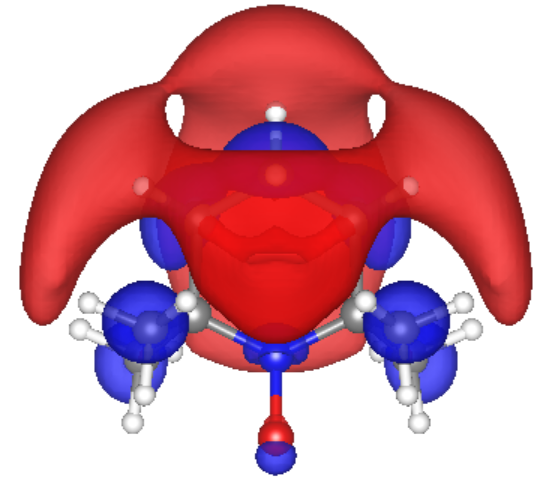
The spin density of the dimer cases consistently show the radical electrons reside primarily at the N-O bonds. In case 5, the Trimer case, the unpaired electrons are delocalized throughout the PTMA wires. Case 5 also indicates hybridization of the molecular orbitals due to the radical electrons’ unique interactions, while the dimer cases resemble two non-interacting monomers, side by side. In the trimer case, the average rate of electron transfer was calculated using Marcus Theory to be 4 X 109 s-1 and the average electron mobility was calculated to be 2.2 x 10-4 cm2 V-1 s-1. These values agree with the reported experimental values for electron transfer in PTMA polymer.
Of the six cases, we found that case 5, representing inter-chain electron hopping, is the best choice for electron transfer. This is because of the hybridization of the radical electrons in the molecular orbitals, and the delocalization of the electrons in the spin density. Our electron transfer rate and electron mobility values for inter-chain hopping are also corroborated by experimental data. A possible area for further exploration would be working on larger systems and other similar configurations to further understand the relationship between the structure and function of PTMA.