News
New paper: Understanding and overcoming biochemical diversity in AL amyloidosis
An invited review for the Israel Journal of Chemistry, to celebrate my mentor Jeff Kelly's receipt of the Wolf Prize:
https://onlinelibrary.wiley.com/doi/10.1002/ijch.202300128
This is a discussion about the parallels and differences between AL and ATTR amyloidosis, and how we have tried to develop tafamidis-like stabilizers for light chains. I hope it's an accessible introduction to the work that we do.
Understanding systemic amyloidosis
This recent article by Amber Dance in Knowable Magazine is a great introduction to systemic amyloidosis. Drawing on interviews with scientists, physicians and patients, it's an overview of where we are, what we've achieved over the last few years, and how much is left to do.
New paper: light chain sequences derived from RNAseq data
Nau et al. Frontiers in Immunology, 14:1167235 2023
We recently published a manuscript describing a method for identifying antibody light chain variable domain sequences from untargeted RNAseq data, which is available from the open-access journal Frontiers in Immunology. This is the result of work by Allison Nau, who started in the lab as a student on the BU MS in Bioinformatics program and continued to work on the project after graduation.
Each patient with AL amyloidosis has a unique involved antibody light chain protein that aggregates to form amyloid fibrils, and a major challenge is to work out why these light chains aggregate but most others do not. One way to do this is to study light chains that do not aggregate despite circulating at high levels, such as those found in multiple myeloma. However, determining light chain sequences is slow and costly. To get around this problem, we asked whether we could extract light chain sequences from existing RNAseq datasets using the program MiXCR.
Our approach worked well enough to apply it to a large set of sequence data from a study by the Multiple Myeloma Research Foundation (MMRF), where hundreds of myeloma patients had their cancer cells sequenced in order to look for factors that influence disease progression and responses to therapy. We were able to identify over 700 light chain sequences from the MMRF data, which is more than twice the number of sequences that we had previously known.
All of these sequences are now deposited in AL-Base, our website that brings together light chain sequences for further study. We are currently in the process of updating AL-Base and analyzing the new data to ask what these new sequences reveal about the factors that lead to amyloidosis.
The work described in this paper was supported by the Karin Grunebaum Cancer Research Foundation and by pilot funding from BU, including an American Cancer Society Institutional Research Grant and the Clinical and Translational Sciences Institute. Allison was also supported by the BU Bioinformatics Program and the BU Genome Science Institute via the Bioinformatics Masters Summer Internship Program.
Welcome Coralie
Coralie Nanfack Tsakem has joined the lab as part of the Boston University STaRS program. Coralie is an undergraduate at The College of New Jersey who will be working in Boston over the summer on mechanisms of AL amyloidosis.
Congratulations to Dylan and Anjaney!
Last month Dylan Dang and Anjaney Shrivastav graduated from the BU Masters in Medical Sciences program after completing their research projects in the Amyloidosis Center.
Dylan's thesis was on the stability and aggregation of LECT2 protein, which is responsible for ALECT2 amyloidosis. Dylan cloned, expressed and analyzed LECT2 in order to study the effect of a disease-associated polymorphism at residue 40, showing that the valine and isoleucine forms of the protein have similar characteristics.
Anjaney wrote his thesis on the ability of small molecules to stabilize antibody light chains and prevent their aggregation. He also cloned, expressed and analyzed proteins in the lab, in this case sequences associated with AL amyloidosis. His work showed that the small molecules that we have identified will stabilize isolated light chain variable domains derived from the IGLV2-14 gene.
Dylan and Anjaney are both applying to medical school this cycle. Congratulations and good luck to both!
Congratulations and goodbye to Alejandra Medina!
We were lucky to have Alejandra Medina Montero join the lab this summer as a scholar on the BU STaRS program. Alejandra's project focused on measuring the ability of small molecules to stabilize antibody light chain variable domains in vitro. She worked hard and had a great time in Boston, and is now back to her undergraduate course at the University of Puerto Rico.

New preprint: LECT2 stability and aggregation
We just posted a manuscript to the bioRxiv preprint server, "An amyloidosis-associated polymorphism does not alter LECT2 stability in vitro". This paper describes work that was carried out in the lab by Liudmila Belonogov and Paris Taylor in 2019 and early 2020, with additional work by Sherry Wong in 2021.
There are many proteins known to aggregate as amyloid fibrils and cause human disease. One of the more recent discoveries is amyloid derived from the LECT2 protein, leukocyte chemotactic factor 2. LECT2 is a signaling protein expressed mainly in the liver, where it has roles in fibrosis and cancer, but its physiological role is not well understood. Amyloid formed from LECT2 has been found in the kidney and liver, in a disease known as amyloid LECT2 (ALECT2) amyloidosis.
The causes of ALECT2 amyloidosis are not known. Unlike inherited forms of transthyretin amyloidosis, there is no obvious mutation that causes disease. Instead, most (or possibly all) individuals are homozygous for a valine (V) residue at position 40 in the LECT2 protein sequence, which can be either valine or isoleucine (I) in the general population. This is not a rare mutation - around a sixth of people have two valine alleles - but we wondered if it might be related to the disease process. Unstable transthyretin proteins are more prone to form amyloid in vitro and in vivo, so we asked whether the V40 variant of LECT2 was less stable than the I40 variant.
We expressed both proteins in E. coli and measured their folding and aggregation using fluorescence spectroscopy. Somewhat surprisingly, we do not see a big difference between the two variants. The valine and isoleucine forms of the protein are similar in every way that we measured. There are some subtle differences in the kinetics of aggregation, but both variants form amyloid at a similar rate. We can't say for certain that there is no difference, but there are no obvious signs that having a valine at position 40 destabilizes LECT2.
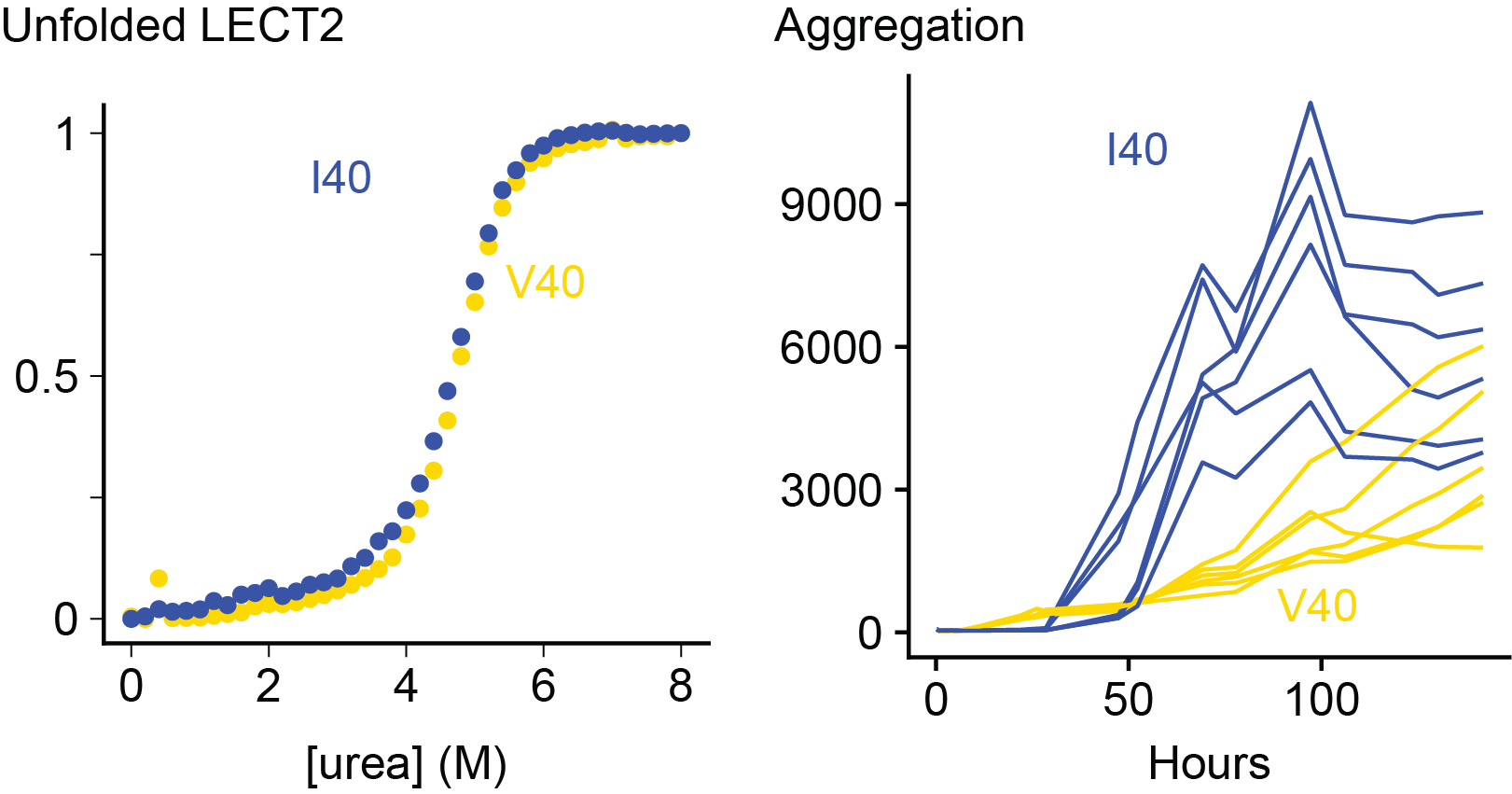
On the other hand, destabilizing LECT2 by removing a structural zinc ion made both variants aggregate much faster. So unfolding of the native state seems to be an important step towards aggregation.
By the spring of 2020, we had managed to get our initial experiments working and collected enough data to show that there are no big differences between the two proteins. However, progress was abruptly halted by the Covid-19 pandemic and we never quite managed to get the experiments restarted properly. With other priorities and funding, I made the decision to put the project on hold for the time being and make our initial data available to anyone who might be interested. It's written into a manuscript to put the data into context, but we have no plans to submit this for peer review until we can better understand what's going on. We're not done with LECT2, but but I would rather have a partial story available to other researchers than let it gather dust until it's "finished".
IDEA award from J&J
We are delighted to be selected for funding from Johnson & Johnson Innovation's IDEA award. This program aims to accelerate the development of new approaches to improving the detection and diagnosis of AL amyloidosis.
Funds from the program will support our research on early diagnosis of amyloidosis. We aim to find out whether antibody light chains that can aggregate as amyloid fibrils are unstable enough that we can identify them in blood samples. This could lead to a test for AL amyloidosis that would identify people at risk, who could benefit from early treatment.
J&J make the only drug approved specifically for AL amyloidosis, the monoclonal antibody daratumumab, so this support for our research should be considered a potential conflict of interest, although the company has no direct influence on the work that we will carry out. We are very grateful for the support.
New paper – Light Chain Stabilization: A Therapeutic Approach to Ameliorate AL Amyloidosis
Morgan, Buxbaum and Kelly; Hemato 2021, 2(4), 645-659
A major goal of our research is to create small molecule drugs to stabilize antibody light chains and suppress their misfolding and aggregation, as a potential therapy for light chain (AL) amyloidosis. Along with my collaborators, Professors Jeff Kelly and Joel Buxbaum at Scripps Research, I wrote a review article about our aims for the project, why we think that it could benefit patients, and our progress to date (open access).
Jeff, who was my postdoctoral advisor at Scripps, recently won the Breakthrough Prize for showing that unstable proteins can cause amyloidosis and that drugs that stabilize these proteins can benefit patients. The outcome of these studies was Pfizer’s drug tafamidis, which is used to treat ATTR amyloidosis, where aggregation of unstable transthyretin (TTR) protein leads to heart and nerve damage. To hear more about the discovery of tafamidis, check out the Scripps Front Row lecture that Jeff gave recently.
It’s worth taking a moment to ask why tafamidis might be considered more significant than the mRNA-directed gene silencing drugs, patisiran and inotersen, which are also approved for treatment of ATTR amyloidosis. To me, the key distinction is that RNA silencing was a therapeutic mechanism looking for a disease, which can be applied to many potential targets. They represent incredible progress, but the strategies were not designed with amyloidosis in mind. TTR has been the target of choice because it’s made in the organ that can be most reliably targeted with the silencers, which is the liver. Whereas tafamidis was designed as a stabilizer of TTR to test a specific therapeutic mechanism with broader implications, that pharmacological stabilization of otherwise unstable proteins could correct genetic disease. One of the components of the cystic fibrosis therapy, trikafta, works in the same way, and there is potential for other disease-associated proteins. So based on this clinical utility of tafamidis for ATTR amyloidosis, we aim to create analogous drugs for AL amyloidosis.
AL amyloidosis treatments are designed to eradicate the clonal plasma cells that secrete amyloidogenic light chains. New therapies do this very well, but many patients continue to experience symptoms from amyloid deposits. For example, only half of patients treated with daratumumab had an “organ response” (improvement in their heart or kidney function) after six months of therapy Part of the reason for this is that amyloid deposits are not cleared by the body, so medicines that aim to remove amyloid fibrils are urgently needed. But this is difficult and clinical data so far is, at best, encouraging rather than spectacular.
Another possible factor is that even low levels of light chain production by surviving clonal plasma cells can continue to drive amyloid formation. We know that more effective eradication of these cells leads to better outcomes, and that long-term remission is possible. So if we can prevent aggregation of whatever light chains remain, either before or after cytotoxic therapy, this should reduce growth of amyloid deposits and allow damaged tissues to recover. We see stabilizers as a complement to existing therapies – stopping production of clonal light chains would still be the priority.
A key conceptual advantage of stabilization is that it acts early in the disease process (the blue inhibition symbol in the figure). We don’t fully understand all of the ways that light chains cause tissue damage, whether as amyloid fibrils, misfolded oligomers or non-native monomers or dimers. But stabilizing the native, folded light chain should prevent formation of all non-native toxic conformations.
We have made some important progress towards drug candidates, but there is still a long way to go. A big milestone is demonstrating that small molecules can bind to light chains and protect them from proteolysis with drug-like, nanomolar affinity. When we set out, we didn’t know if would be possible at all.
Importantly though, there are a number of questions that we can’t answer without clinical trials, so we need to be cautious about how we approach treating patients. There are no animal models that recapitulate all of the features of systemic amyloidosis, so we can’t look for efficacy in mice. We need to make sure that our molecules are as effective as we can make them in lab assays – ensuring that they bind tightly and specifically to light chains, and that they are non-toxic. But we will need a clinical trial in humans to learn whether the molecules are effective enough, and whether stabilizing light chains actually suppresses amyloid formation.
We are confident that we can make a molecule that is safe for people, and stabilizes light chains. And we are cautiously optimistic that stabilization will suppress amyloid formation and be of benefit to patients. Let’s see how far we can take this idea.
AL-Base update 9/2021
We recently updated and relaunched our database of antibody light chain sequences, AL-Base. This unique collection of curated protein sequences is a useful resource that has been used by many groups around the world. The database and site were originally built by Kip Bodi in the Amyloidosis Center in 2008, but had been due for an update. We had been collecting new sequences and trying to work out how to update the site when its server went offline earlier this year. Unfortunately we were unable to resurrect the site and had to rebuild it from scratch (although we had at least managed to back up the data).
We reached out to the School of Public Health's Biostatistics and Epidemiology Data Analytics Center (BEDAC), and Dr. Axin Hua has built a new site, on a modern framework, that has restored AL-Base's core functionality. The site has a new address at https://wwwapp.bumc.bu.edu/BEDAC_ALBase but the old URL (albase.bumc.bu.edu) will redirect there. The site still looks very similar but the underlying web application is completely new. For example, downloading sequences should now be much easier than in the previous version. There are still some things to rebuild, but the database is usable again. Looking ahead, we aim to extend the database and add new functionality over the coming months.
There are some changes to the sequences available. We have removed a number of duplicated or redundant sequences, so each sequence is now unique. This means that there are fewer sequences in total, but analyzing these sequences should be easier. We are working on a way to handle multiple instances of the same sequence, such as when there are both nucleotide and protein sequences in various databases. In the meantime, we have tried to be conservative and excluded all copies of a sequence if there is any ambiguity about which is the "correct" one.
We have also scoured the literature for new light chain sequences associated with amyloidosis or related plasma cell disorders, adding 177 new sequences. In total, AL-Base now contains 556 unique AL-associated light chains and 242 sequences from other plasma cell disorders.
Please let us know if you use the new site and have any suggestions for improvements or features that you would like to see. If you use AL-Base in your work, please cite the original paper and acknowledge the support of the NIH grant, HL68705.