News
New paper: Structural Basis for the Stabilization of Amyloidogenic Immunoglobulin Light Chains by Hydantoins
How does a curved molecule fit into a flat binding pocket?
Structural basis for the stabilization of amyloidogenic immunoglobulin light chains by hydantoins.
Nicholas L. Yan, Diogo Santos-Martins, Enrico Rennella, Brittany B. Sanchez, Jason S. Chen, Lewis E. Kay, Ian A. Wilson, Gareth J. Morgan, Stefano Forli, Jeffery W. Kelly
Bioorganic & Medicinal Chemistry Letters
Volume 30, Issue 16, 15 August 2020, 127356
When we identified small molecules that can stabilize antibody light chains, aiming to develop a drug for AL amyloidosis, most of the hits from our screen were flat molecules. The molecule that we investigated most thoroughly, coumarin 1, has a two-ring aromatic core that slides in between two domains of the light chain dimer. The overall binding pocket, which is not present in the unbound light chain structure, is deep and flat. While this allows for reasonably tight binding, flat, hydrophobic molecules are generally not ideal as drug candidates because they tend to bind to many different proteins. Coumarins are generally regarded as “promiscuous” in this sense. We therefore had a potentially long road ahead in order to design a stabilizer molecule that binds tightly and specifically to light chains. Our new paper describes our first step on that road.
Of the twenty or so molecules that came out of our initial screen, two related molecules are much less flat than the others. Both contain a 5-membered hydantoin ring at their core, linked to a trifluoromethylated phenyl ring. We called them coumponds 7 and 8. It was not obvious how these molecules could bind to the light chain, even though we had competition binding data that was consistent with them binding at the same site as the coumarins.
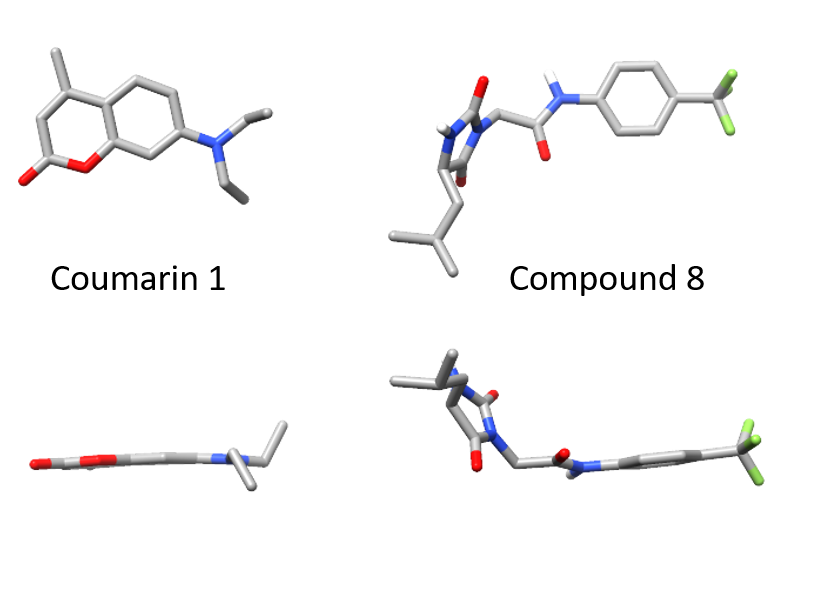
I’d previously met Stefano Forli at Scripps happy hour and we’d talked about doing some computational modeling of potential stabilizer molecules. (Go meet your colleagues, folks!) Stefano works on computational drug design and in silico modeling. Stefano’s postdoc, Diogo Santos-Martins, was able to carry out computational docking experiments that suggested a potential binding conformation of the hydantoin molecules. The phenyl ring and its trichloromethyl group packs into the known coumarin binding site, but the hydantoin and the rest of the molecule stick out, interacting with new residues on the surface of the light chain.
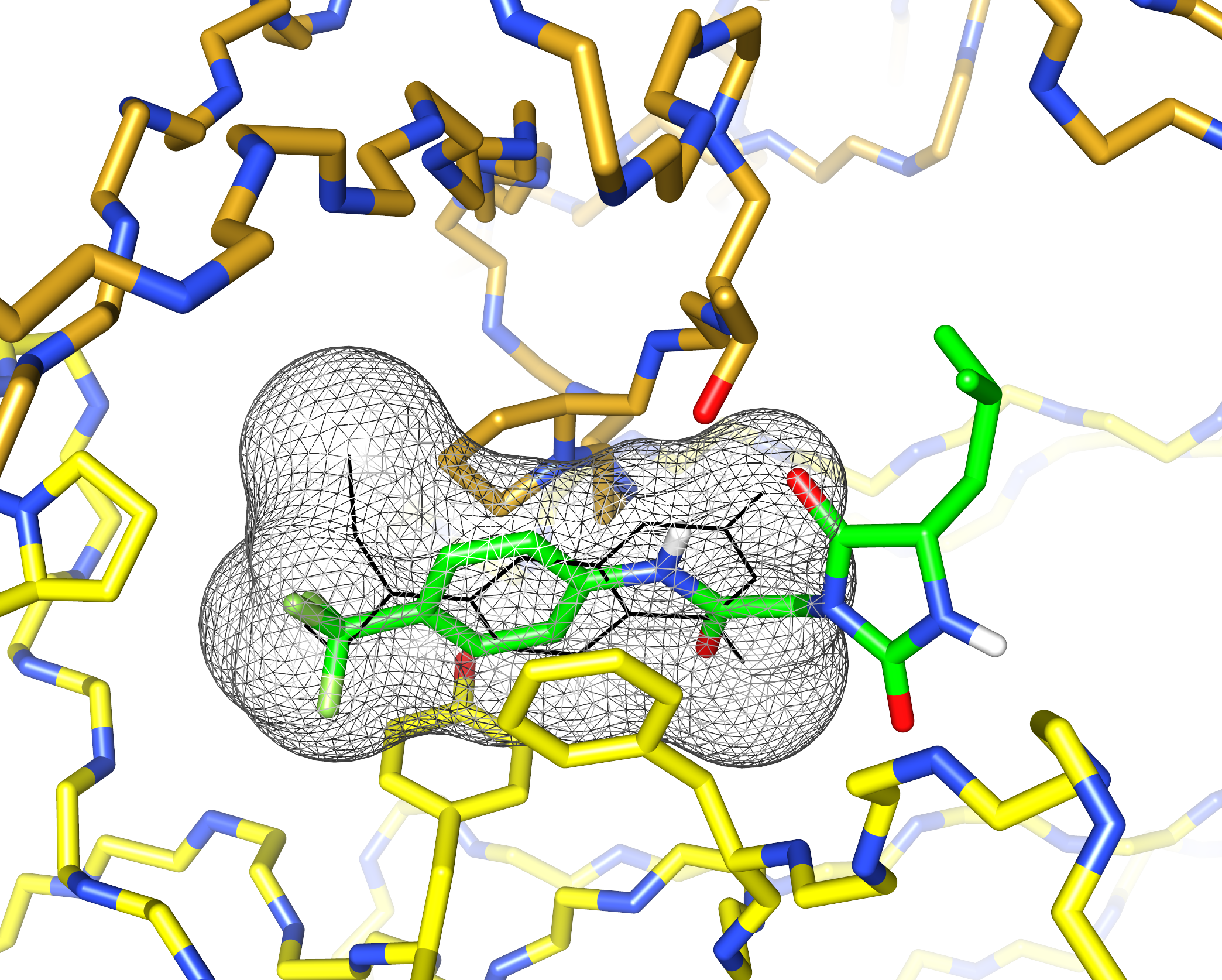
This was exciting because increasing the number of interactions between a protein and its ligand is a good way to improve affinity and specificity. Nick Yan, a graduate student working with me in the Kelly lab at Scripps, set about making derivatives of these molecules and managed to crystalize one of the complexes. The X-ray structure showed almost exactly the same binding conformation as the computational model. We verified that this also happens in solution, using NMR and analytical ultracentrifugation.
One difference was in the stereochemistry of the structure. Nick used a racemic mixture of the two forms of the hydantoin molecule in his experiments, but only one form was visible in the crystal. Diogo showed that this enantiomer bound more tightly in the simulations. Nick separated the two forms and found that one was able to protect light chains from proteolysis more effectively than the other.
This paper is a neat demonstration of how computational tools can help to prioritize screening hits. The new interactions that Diogo’s work pointed out have allowed Nick to design and synthesize more effective small molecule stabilizers. That work is still in progress, but we’re moving in the right direction.
This link has free access to the full paper until August 2020, but might need you to suspend your pop-up blocker: https://authors.elsevier.com/c/1bIAs,LsIFmv4Y
A high-throughput screen for protein stability
We have developed a new method for measuring protein stability that can be used in high throughput screens. We hope that this will be a useful alternative to existing methods for researchers looking for molecules that bind to and stabilize proteins. Details are in our new paper.
Proteins are dynamic, flexible polymers that can form a range of structures. In cells, proteins are synthesized on ribosomes, acquire their functional shape (which may or may not be folded) and are eventually degraded when they are no longer needed. A new generation of drugs designed to alter the stability and lifespan of proteins is expanding the definition of a “druggable” target and making a difference in many patients’ lives. At one end of this spectrum are drugs that result in the degradation of proteins, which work by increasing the rate at which proteins are sent to the proteasome within cells. At the other end are stabilizers and pharmacological chaperones, which increase the production and lifespan of proteins that are prone to misfolding. We are working with collaborators at Scripps Research to develop molecules that stabilize antibody light chains and prevent their aggregation in AL amyloidosis.
However, rationally designing small molecules that modulate protein structure and stability, rather than enzyme activity, is difficult. Stability can mean an individual protein’s lifespan or its propensity to maintain its folded structure. These properties are intimately linked because proteins are generally removed by proteases, which preferentially degrade unfolded proteins. To measure stability in an isolated protein, we typically put it under conditions that perturb the folded state, then ask how much folded protein remains. This could be by raising the temperature, changing the pH, adding protease to digest the unfolded protein or other methods. The difficulty lies in distinguishing folded from unfolded protein. This is possible using spectroscopy or size separation, depending on how the protein was perturbed. When we measure the stability of light chains, we use fluorescence emission from the protein’s tryptophan residues or electrophoresis to separate intact from proteolysed protein.
Neither of these methods scales to the throughput needed to screen a large library of potential drug molecules – you can’t easily make the measurements on a 1536-well plate. One method that can be used is called differential scanning fluorimetry, or ThermoFluor. Here, proteins are heated until they begin to unfold. Hydrophobic dye molecules can bind to partially folded proteins, enhancing the fluorescence of the dye. As the temperature increases, dye fluorescence increases and then eventually decreases again as the protein unfolds completely. The peak of the resulting curve occurs at a protein’s thermal denaturation (“melting”) temperature. A molecule that stabilizes the protein will cause an apparent increase in the melting temperature. This can work well, but like all assays, it’s prone to interference.
We looked at using a ThermoFluor assay to find small molecules that stabilize light chains, but the initial results were not promising. So we tried another approach. Proteolysis depends on stability, so we looked for a way to ask whether a protein molecule was intact or had been cleaved by protease. We labeled light chain molecules with fluorescein, a simple fluorescent dye, and measured their fluorescence polarization as they were digested by protease.
Fluorescence polarization (also known as fluorescence anisotropy) measures rotational diffusion – the rate at which molecules tumble in solution. When a molecule absorbs light, there is a delay before that light is reemitted as fluorescence. If the molecule rotates in that time period, the polarization of the emitted light differs from that of the absorbed light. Because the molecules tumble randomly, the net polarization is averaged out if the delay between absorbance and emission is longer than the time it takes for the molecule to rotate. Small molecules tumble rapidly and larger molecules tumble more slowly, so the polarization of emitted light is averaged out more rapidly by small molecules than by large ones. This fluorescence polarization measurement can be made quickly and easily using a platereader.

Josh Blunden, who was a summer student in the Kelly lab, was able to get this assay working. The fluorescence polarization of light chains decreases as they are degraded by protease, and the rate of degradation depends on the protein’s stability. Therefore, we could run the proteolysis reactions in parallel with different potential stabilizer molecules (650,000 in this case) to carry out our screen. We call this method the “protease-coupled fluorescence polarization”, or PCFP, assay. We found 2,777 hits that were active in the screen. Like all screens, there are ways for this to go wrong. We saw interference with the assay that seems to be from binding of dye or protein to the plate surface, but this could be solved by adding detergent or using low-binding plates. An obvious failure mode here is that the small molecule could inhibit the protease, rather than stabilize the light chain. To identify and remove any of these false positive molecules, we repeated the screen but used a different protease with a distinct catalytic mechanism (thermolysin, rather than the proteinase K that we used in the main screen). Several molecules had no effect in this assay, suggesting that they were false positive hits. One molecule was a well-characterized protease inhibitor drug, bortezomib.
After further rounds of validation and counterscreening, we are confident that the molecules described in the paper are genuine stabilizers of light chains. We are working to create more effective molecules that could become drug candidates.
We think that our PCFP method could be useful for other researchers who are looking for ways to measure stability in high throughput screens. In theory, any binding event that perturbs protease sensitivity can be measured this way. It has some advantages and disadvantages compared to a ThermoFluor assay, so it may work better in some cases. The PCFP assay is a single read in a polarization-capable platereader, whereas a ThermoFluor assay requires a long process in a qPCR instrument. PCFP may be less prone to artifacts such as protein aggregation, evaporation or dye displacement. However, it requires site-specific labeling of the protein, which can be difficult to optimize, whereas a ThermoFluor assay uses unmodified protein. The chemistry and optical readout are distinct from the ThermoFluor assay, so the two methods should be complementary. This means that true positive hit molecules should be active in both assays, while false positives may be more specific to one or other assay. One useful property is that the assay works at low protein concentration, which can be an advantage for screening. We were able to run the whole screen using 10 mg of fluorescein-labeled protein.
There’s also the potential to use this assay, or something similar, as a function-agnostic way to find protein binding molecules. Most binding assays require knowledge about the binding site, a known ligand or some kind of functional assay to work. For example, it’s easy to screen for protease inhibitors using fluorogenic substrates, or by looking for molecules that will displace a known inhibitor. Alternatively, cell-based assays that measure the activity of a signaling pathway or reporter gene have been used for all kinds of screens. One potential advantage of the PCFP assay is that you don’t need to know how the target protein works, or where its ligands bind (even its exact structure, as long as you can attach a fluorophore). The one thing you do need is data suggesting that the proteolysis of the target protein is stability-dependent, which wouldn’t necessarily be true for an intrinsically disordered region, for instance. It might be possible to get around that by using a more specific protease. We plan to try similar assays with other systems in order to see how far we can take the idea.
It’s a little surprising that no-one seems to have tried this before. Or maybe they have, but it didn’t work in their system and it was never published. I really hope that this method can solve someone’s problem and help move a project forwards. Please let me know if you want to try it and would like some advice.
New paper: Small molecules that stabilize light chains
A major goal of the lab is to develop new therapies for amyloidosis. Today, our paper describing a big step towards a drug for AL amyloidosis was published. We have identified small molecules that bind to antibody light chains and prevent them from unfolding, which could potentially reduce amyloid formation in patients. We’re a long way from a drug yet, but this paper shows where and how a drug might bind. One of the molecules is a simple fluorescent dye that could be a useful tool for further studies. Along the way, we figured out a new way to screen large libraries of molecules to find stabilizers. This was a big team effort, mostly carried out at Scripps Research during my time as a postdoc there, and I especially want to thank co-authors Jeff Kelly and Nick Yan, who put a huge amount of work into making this happen.
(I do have a conflict of interest here: we have applied for a patent for this work and there’s a chance that I might make some money from sales of an eventual drug.)
AL amyloidosis can be treated by killing the clonal plasma cells that secrete the antibody light chains that aggregate to cause disease. However, the treatments are very harsh and don’t work for everyone. If we can prevent the misfolding of light chains, independently of the cells that produce them, we may be able to stabilize patients and improve their quality of life. Ideally, this strategy would allow sick patients to tolerate chemotherapy.
Amyloidosis can be caused by destabilization of the precursor protein, which encourages the structural changes that lead to aggregation. In transthyretin amyloidosis, this is caused by hereditary mutations or ageing. Stabilizing transthyretin using small molecules (tafamidis, diflunisal or AG10) prevents misfolding and can prevent disease progression. In AL amyloidosis, each patient has a different light chain sequence but there’s evidence that less stable light chains are more likely to cause disease. Our work on full-length light chains suggested that stabilizing these proteins, rather than the isolated variable domains that most other groups had focused on, could be helpful.
The Kelly lab at Scripps developed tafamidis as a stabilizer of transthyretin, so we have some experience with this kind of thing. Transthyretin is a hormone transport protein with a deep binding pocket for its natural ligand, thyroxin. Unfortunately, light chains don’t have known ligands or any obvious binding pockets, so we needed to start from scratch.
The standard way to find drugs for an unknown target is to use a high-throughput screen. Here, a measurement is repeated thousands of times in parallel, with a different small molecule in each reaction. The key to successful screening is to start with a large library of small molecules, which means robotic liquid handling and 1,536-well plates. Screening for compounds that enhance stability is difficult, because the methods used in most laboratories can’t be used on microwell plates.
We developed a method to get around this based on proteolysis, using fluorescence polarization as a readout. Josh Blunden, an undergraduate student who worked in the lab in the summer of 2016, did a lot of the legwork needed to get this started. Then Steve Brown at Scripps helped turn it into a working assay that we could use to screen the compound library at Scripps Florida. The assay works well and it could be useful for other targets. Somewhat surprisingly, no-one seems to have done this before. We’re intending to publish a methods paper to help other groups use it.
This is where my part in the story could have ended. High throughput screening is expensive (we used a thousand or so plates, at five dollars each, for example). Getting a grant to fund the screen might have taken a year or more. Fortunately, Jeff was sufficiently enthusiastic about the prospects of developing a drug that he was able to scrape the money together and say “Make it so!” It’s a huge commitment that wouldn’t have been possible in most labs and I’m really grateful that we were able to go for it.
Tim Spicer and Virneliz Fernandez-Vega at Scripps Florida screened 650,000 compounds and identified 2,777 hits. Screens tend to throw up a lot of false positives, so we used secondary screens to narrow the list down to 128 molecules that they shipped to us. We picked out the best 16 molecules, which could be divided into four chemical classes. Finally, we chose one molecule for detailed characterization.
What’s so special about the final molecule? Part of the answer is that we had a bottle of it in the cupboard – it’s a commercially-available fluorescent dye called coumarin 1. All the best molecules had similar activity, so we picked this one because it was convenient and readily available to other researchers. We do have some evidence that the other molecules bind in the same way, so we were able to use coumarin 1 as a tool to ask about the mechanisms and consequences of stabilizer binding. We verified that coumarin 1 and the other small molecules stabilize multiple light chains against different types of perturbations, so we’re confident that it works in the way we were aiming for.
At the end of 2017, Nick Yan joined the lab as a graduate student. He quickly took on the project of characterizing the small molecule hits from the screen. With the help of David Mortenson in the Kelly lab, and Ian Wilson’s structural biology group at Scripps, Nick solved two crystal structures: a light chain protein alone, and the same light chain in the presence of coumarin 1. The structures show where the small molecule binds – between the two variable domains of the light chain dimer. We verified that light chains bind to coumarin 1 in the same way in solution using NMR, in collaboration with Enrico Rennella and Lewis Kay in Toronto, who have collaborated with us to understand the dynamics of the light chain dimer.
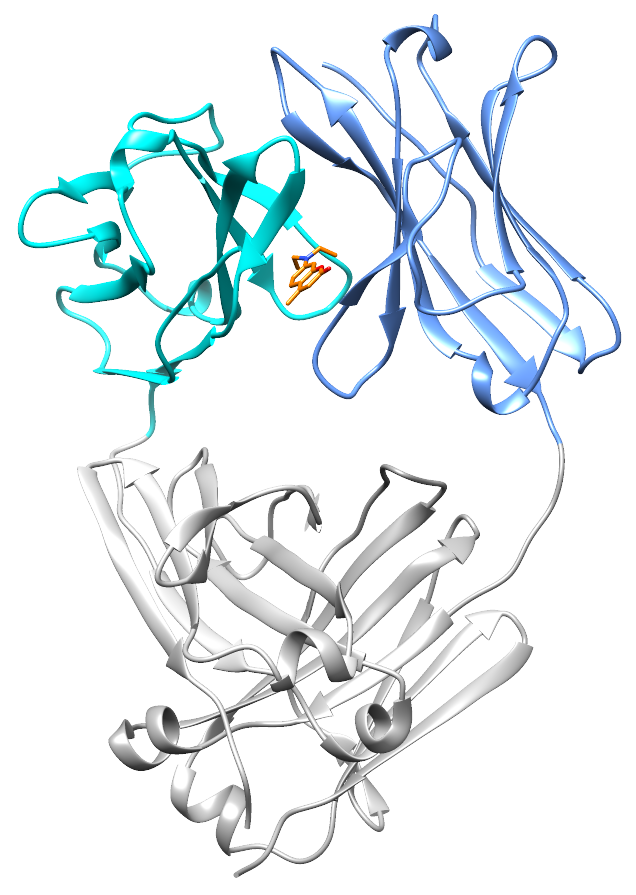
What’s really encouraging and unexpected about the binding site is that it’s built from residues that are very similar between different light chains. Each AL amyloidosis patient has a unique protein sequence, so a potential drug needs to bind at a site that’s conserved between different light chains. That’s what we found – the binding site is made up of residues that form the structural core of the variable domains. The same residues are present in more than 90% of all light chains. Coumarin 1 binds into a pocket that’s not present in the unliganded native structure, a so-called “cryptic” binding site. Importantly, the binding site is not present in normal antibody heavy chain-light chain dimers, which means that the molecules should be specific for free light chains.
Coumarin 1 is not an ideal drug molecule. It doesn’t bind tightly enough to the light chain, and it will probably stick to a lot of other molecules in the body. We’re more excited about the binding site that we discovered. We will make molecules that bind much more tightly and specifically to light chains, based on the structure of the complex.
On the other hand, coumarin 1 is a fluorescent dye, and its fluorescence gets much brighter when it’s bound to light chains than when it’s free in solution. This means that we can use it as a probe for folded light chain dimers. This is not something that’s written on the bottle and it wouldn’t necessarily have been an obvious thing to try. But this “fluorogenicity” has been a big part of the lab’s research and it’s such a useful property that we routinely check for it in new compounds. In the paper, we use this property to ask how tightly the small molecule binds to different light chains, and whether other molecules bind in the same site. We hope that this will be a useful tool for other amyloidosis researchers.
I’ve spent three years working on this project and it’s great to see it published at last. It’s really the beginning of a process that we hope will lead to a drug for what is often an untreatable disease. There are a lot of challenges before we can think about clinical trials, but I’m confident that we will be able to make progress.
CryoEM structures of AL amyloid fibrils
Two manuscripts were published last week describing high-resolution 3D structures of light chain amyloid fibrils extracted from patient heart tissue. Swuec and coworkers solved the structure of a fibril derived from a lambda 6-57 light chain using cryo electron microscopy (cryoEM); Radameker and coworkers did the same for a lambda 1-44 light chain. These structures provide important information about how amyloid forms in patients. However, the structures themselves are remarkably different, which shows just how much more we have to learn.
Amyloid fibrils are large and non-crystalline, which makes them difficult to study using the structural techniques developed for soluble proteins. So although there have been some important insights, the detailed molecular structures of amyloid fibrils from diseased tissue have remained out of reach. That changed in 2017 with the publication of structures of tau filaments from the brain of a patient who had died of Alzheimer’s disease. A series of technological advances in EM hardware and software over the last few years has enabled this work. It’s great to see it applied to AL amyloidosis. If you’re interested in the really diving into the details of cryoEM, Caltech has a free, detailed online course.
Both fibrils were extracted from heart tissue using repeated washes with water and buffer to remove non-amyloid material. Amyloid is pretty tough and survives multiple rounds of homogenization in a blender. Isolated fibrils were suspended in an ultrathin film of water, then rapidly frozen in liquid ethane. The fast freezing prevents ice crystals forming, which would damage the fibrils.
Structure determination by EM relies on the repetitive nature of the fibrils. Gigabytes of EM images are analyzed to identify individual fibrils, which are then sliced into segments. Each segment is a projection of the fibril, rotated according to its helical twist. By combining many, many segment images, it’s possible to reconstruct the 3D structure that created the projection. Ten years ago, the best reconstructions were intricate blobs that showed where the protein was, but not which bit of protein was where. The new generation of structures can identify specific sidechains and their interactions.
Both fibrils consist of a single protofilament, formed from residues of the variable domain. As has been observed in other fibrils, the constant domain has been partly removed, presumably by proteolysis. Parts of both protein chains are missing from the resolved density, which probably means that they are less well-structured than the resolved core of the fibrils. Both structures have a complex, non-native arrangement of β-strands that makes up the core of the fibril. The native disulfide bond is present in the core of the structures, but the rest of the protein chains are arranged into a series of parallel, in-register cross-beta structures, where the hydrogen bonds run along the long fibril axis. Here are the lambda 1-44 fibril and the lambda 6-57 fibril depicted as topology diagrams.
This non-native structure indicates that the V-domain must unfold in order to form aggregates, consistent with a role for reduced LC stability. This is highlighted in the lambda 1-44 fibril because the orientation of the beta strands around the disulfide are reversed relative to their orientation in the native state of the fibril. Importantly, the complementarity-determining regions of the V-domain pack into a complex structure within the fibril core. This indicates that different LC sequences will alter the stability of the fibril core, which may also constrain which patient LC sequences are able to form amyloid in vivo.
Beyond these similarities though, the structures are remarkably distinct. The regions of beta structure and the overall packing of the cross-beta elements are different. One N-terminus is buried right in the center of the fibril, while the other is unstructured. (This highlights the importance of using native-like termini in biochemical studies - would a his-tagged version of this protein form the same structures?) The lambda 6-57 fibril does not have resolved density for residues 37-65, but these are structured in the lambda 1-44 fibril.
To get a better sense of how different the structures are, I aligned the sequences of the two light chains and looked at where the beta strands are in the native proteins and the fibrils. (See the figure - I took the data from the figures in the papers.) The sequences are 63% identical and the native beta structures are essentially the same as each other. But the structured regions of the fibril and the patterns of beta strands – the arrows above the sequences – are very different.
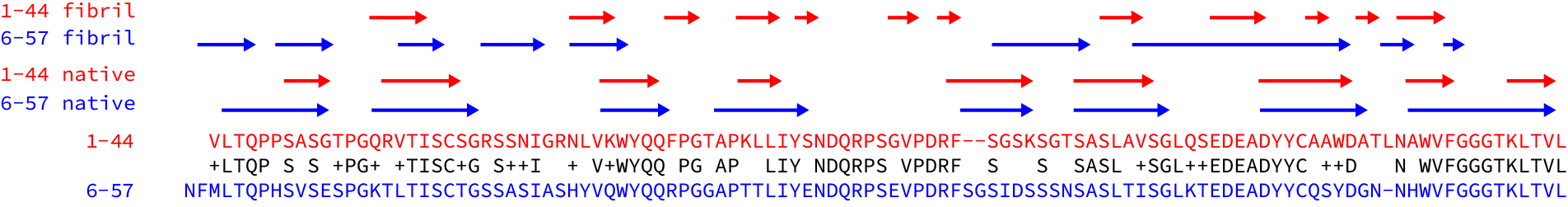
These differences raise a lot of questions. Will every amyloid fibril be unique? Can the same light chain form different structures? Are fibrils in different tissues more, or less similar than fibrils from different patients? Is the structure of a fibril determined primarily by its sequence, or are interactions with other molecules important? Are kappa fibrils different to lambda fibrils?
What’s also interesting is what the structures don’t show. There’s almost no trace of native-like structure in either fibril, which means that the native light chain probably has to unfold completely in order to aggregate. This rules out a large class of models for fibrils that retained some of the native structure. In addition, there’s nothing that would obviously cause toxicity. And there’s very little in common between the fibrils that could explain why they cause organ dysfunction in similar ways.
As ever, there’s plenty more work to be done. But these structures represent an important milestone. It’s really helpful to have a detailed model of what a structure might look like in order to ask what different parts of it might be doing. Congratulations to both sets of authors!
Pilot grant award for plasma cell genomics
Great news - we've been awarded a pilot grant to study gene expression in AL amyloidosis patient-derived plasma cells, and to sequence circulating tumor DNA from these patients' blood. The funding comes from the American Cancer Society via an institutional grant to BU.
Our long-term goal is to ask how genetic changes in plasma cells lead to disease, and whether we can identify specific changes that could help select the most effective drugs for individual patients. The first step is to set up the lab to carry out these experiments and test out the procedures, looking for ways to get the best data from patient samples. We will be sequencing mRNA from plasma cells to measure gene expression, and cell-free DNA from blood to ask whether we can measure the number of cancerous cells in the bone marrow. We can make use of methods developed for studying multiple myeloma, a more aggressive plasma cell cancer - and maybe help to understand more about that disease, too.
This is a great opportunity to establish some complex experimental systems in the lab. We don't yet know exactly what will work, so this pilot grant allows us to test out some approaches and acquire some preliminary data before we look for funding for a bigger project.
I'm really grateful to the Cancer Center and the ACS for this opportunity. I'm confident that we'll be able to turn their generosity into some important insights into the biology of amyloidosis.
New paper: Domain interactions in light chains
Our paper using NMR to study the dynamics of antibody light chains has just been published online. We collaborated with Enrico Rennella and Lewis Kay at the University of Toronto, who are experts in NMR spectroscopy, to ask how forces within and between the domains of a full-length light chain dimer contribute to the stability of the overall protein. We found that the dimer is more stable than its component domains, showing that there's a strong cooperativity. This coupling of folding and dimerization helps to explain why light chain dimers are much more resistant to aggregation than their component domains. Our results suggest why AL amyloidosis is so rare -- the dimeric structure of the light chain prevents the aggregation-prone regions from forming amyloid fibrils. More
Hello world!
I recently arrived in Boston and started at the Boston University School of Medicine, where I'm building a research program at the Amyloidosis Center. This website will describe our research.