Interdisciplinary Team Sheds Light on How Proteins Bind
Finding Could Open Up New Drug Discovery Opportunities
By Mark Dwortzan
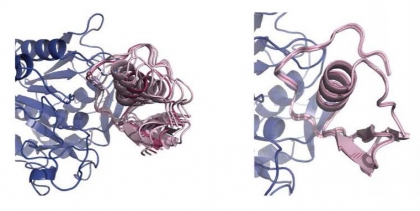
“Transient encounter complexes” of an enzyme produced by a bacterium present in the human gut binding to a fragment of a protein in turkey eggs which inhibits the enzyme.
Over the past six years, an interdisciplinary team of College of Engineering faculty members—Professor Sandor Vajda (BME, SE), Research Assistant Professor Dima Kozakov (BME), Professor Yannis Paschalidis (ECE, SE) and Associate Professor Pirooz Vakili (ME, SE)—have been developing a set of powerful optimization algorithms for predicting the structures of complexes that form when two proteins bind together—structures that, in some cases, generate erroneous cell signaling pathways that can trigger cancer and other inflammatory diseases.
Incorporated into Vajda’s and Kozakov’s protein-protein docking server ClusPro—a website to which any user can submit the three-dimensional coordinates of two proteins and receive a supercomputer-calculated prediction of the structure of the complex formed by those proteins—these algorithms have enabled more than 3,000 research groups across the globe to better understand the inner-workings of the cell and explore potential drug targets without having to run expensive, time-consuming lab experiments.
Now the research team behind these algorithms has, through lab experiments and computational analysis, obtained a sharper understanding of how two proteins come together to form a complex, and plans to apply that knowledge to boost the speed and accuracy of ClusPro’s predictions. They and collaborators from the Hebrew University of Jerusalem and the National Institutes of Health (NIH) report on this new development in a new article in eLife, an open source journal for outstanding biomedical research.
A joint effort of Boston University’s Center for Information and Systems Engineering and Biomolecular Engineering Research Center supported by a five-year, $1.6 million grant from the NIH, the project combines Paschalidis’ and Vakili’s expertise in optimization and systems theory with Vajda and Kozakov’s knowledge of biophysics and bioinformatics.
“The research was a beautiful combination of physics with mathematics,” said Paschalidis. “We leveraged techniques popular in control systems developed to describe movement of complex 3-D objects, such as a robot arm, as well as machine learning methods used to analyze large data sets.”
“Preventing proteins from binding to the wrong partners is an increasingly prominent concept in drug design,” said Janna Wehrle, PhD, of the NIH National Institute of General Medical Sciences, which partially funded the research. “These new computational methods developed by the Boston University team will help researchers quickly discover both healthy protein pairs and disease-causing pairs that we might want to break up.”
Until now, scientists were unable to characterize how protein-protein complexes form from two individual proteins—each analogous to a distinctly-shaped Lego block—because their interactions from the moment they come in contact to the moment they “snap into place” were too fast to detect. But an emerging nuclear magnetic resonance (NMR) technique has made it possible to track their rapidly changing configurations from rendezvous to docking using radio waves.
Applying this technique, the College of Engineering team determined that its protein-protein docking algorithms were already generating these exact transitional states, but labelling them as “false positives” alongside the correctly identified final protein-protein complex.
“What we have so far been calling false positives are ‘transient encounter complexes,’ temporary structures the proteins form as they ‘search’ for the one orientation that will enable them to bind successfully,” said Paschalidis.
All protein-protein encounter complexes are characterized by low energy, with the lowest energy expected to occur at the final, stable complex. By systematically analyzing the energy values corresponding to the transient complexes, the researchers found that with each successive interaction, the intersecting proteins have fewer and fewer ways to twist and turn, thereby accelerating their path to binding. This explains how two proteins can dock very quickly despite the many nooks and crannies that must line up to seal the deal.
The College of Engineering team next aims to exploit its findings to make its docking algorithms faster and more accurate. The researchers also plan to examine the implications of their work for protein-DNA and protein-small molecule interactions that are important in genetic regulation and drug discovery, respectively.
See movie of transient protein-protein encounter complexes.