New paper: Small molecules that stabilize light chains
A major goal of the lab is to develop new therapies for amyloidosis. Today, our paper describing a big step towards a drug for AL amyloidosis was published. We have identified small molecules that bind to antibody light chains and prevent them from unfolding, which could potentially reduce amyloid formation in patients. We’re a long way from a drug yet, but this paper shows where and how a drug might bind. One of the molecules is a simple fluorescent dye that could be a useful tool for further studies. Along the way, we figured out a new way to screen large libraries of molecules to find stabilizers. This was a big team effort, mostly carried out at Scripps Research during my time as a postdoc there, and I especially want to thank co-authors Jeff Kelly and Nick Yan, who put a huge amount of work into making this happen.
(I do have a conflict of interest here: we have applied for a patent for this work and there’s a chance that I might make some money from sales of an eventual drug.)
AL amyloidosis can be treated by killing the clonal plasma cells that secrete the antibody light chains that aggregate to cause disease. However, the treatments are very harsh and don’t work for everyone. If we can prevent the misfolding of light chains, independently of the cells that produce them, we may be able to stabilize patients and improve their quality of life. Ideally, this strategy would allow sick patients to tolerate chemotherapy.
Amyloidosis can be caused by destabilization of the precursor protein, which encourages the structural changes that lead to aggregation. In transthyretin amyloidosis, this is caused by hereditary mutations or ageing. Stabilizing transthyretin using small molecules (tafamidis, diflunisal or AG10) prevents misfolding and can prevent disease progression. In AL amyloidosis, each patient has a different light chain sequence but there’s evidence that less stable light chains are more likely to cause disease. Our work on full-length light chains suggested that stabilizing these proteins, rather than the isolated variable domains that most other groups had focused on, could be helpful.
The Kelly lab at Scripps developed tafamidis as a stabilizer of transthyretin, so we have some experience with this kind of thing. Transthyretin is a hormone transport protein with a deep binding pocket for its natural ligand, thyroxin. Unfortunately, light chains don’t have known ligands or any obvious binding pockets, so we needed to start from scratch.
The standard way to find drugs for an unknown target is to use a high-throughput screen. Here, a measurement is repeated thousands of times in parallel, with a different small molecule in each reaction. The key to successful screening is to start with a large library of small molecules, which means robotic liquid handling and 1,536-well plates. Screening for compounds that enhance stability is difficult, because the methods used in most laboratories can’t be used on microwell plates.
We developed a method to get around this based on proteolysis, using fluorescence polarization as a readout. Josh Blunden, an undergraduate student who worked in the lab in the summer of 2016, did a lot of the legwork needed to get this started. Then Steve Brown at Scripps helped turn it into a working assay that we could use to screen the compound library at Scripps Florida. The assay works well and it could be useful for other targets. Somewhat surprisingly, no-one seems to have done this before. We’re intending to publish a methods paper to help other groups use it.

This is where my part in the story could have ended. High throughput screening is expensive (we used a thousand or so plates, at five dollars each, for example). Getting a grant to fund the screen might have taken a year or more. Fortunately, Jeff was sufficiently enthusiastic about the prospects of developing a drug that he was able to scrape the money together and say “Make it so!” It’s a huge commitment that wouldn’t have been possible in most labs and I’m really grateful that we were able to go for it.
Tim Spicer and Virneliz Fernandez-Vega at Scripps Florida screened 650,000 compounds and identified 2,777 hits. Screens tend to throw up a lot of false positives, so we used secondary screens to narrow the list down to 128 molecules that they shipped to us. We picked out the best 16 molecules, which could be divided into four chemical classes. Finally, we chose one molecule for detailed characterization.
What’s so special about the final molecule? Part of the answer is that we had a bottle of it in the cupboard – it’s a commercially-available fluorescent dye called coumarin 1. All the best molecules had similar activity, so we picked this one because it was convenient and readily available to other researchers. We do have some evidence that the other molecules bind in the same way, so we were able to use coumarin 1 as a tool to ask about the mechanisms and consequences of stabilizer binding. We verified that coumarin 1 and the other small molecules stabilize multiple light chains against different types of perturbations, so we’re confident that it works in the way we were aiming for.
At the end of 2017, Nick Yan joined the lab as a graduate student. He quickly took on the project of characterizing the small molecule hits from the screen. With the help of David Mortenson in the Kelly lab, and Ian Wilson’s structural biology group at Scripps, Nick solved two crystal structures: a light chain protein alone, and the same light chain in the presence of coumarin 1. The structures show where the small molecule binds – between the two variable domains of the light chain dimer. We verified that light chains bind to coumarin 1 in the same way in solution using NMR, in collaboration with Enrico Rennella and Lewis Kay in Toronto, who have collaborated with us to understand the dynamics of the light chain dimer.
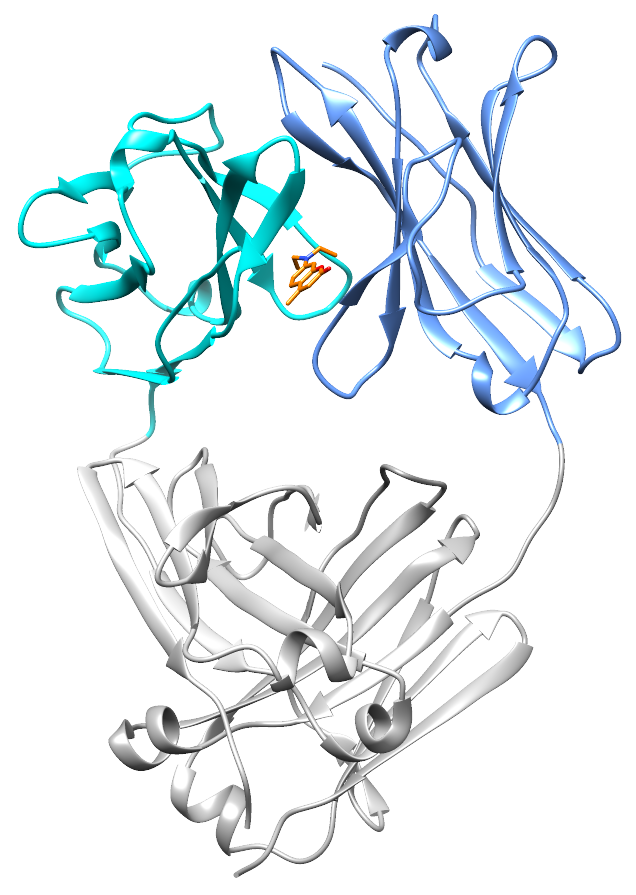
What’s really encouraging and unexpected about the binding site is that it’s built from residues that are very similar between different light chains. Each AL amyloidosis patient has a unique protein sequence, so a potential drug needs to bind at a site that’s conserved between different light chains. That’s what we found – the binding site is made up of residues that form the structural core of the variable domains. The same residues are present in more than 90% of all light chains. Coumarin 1 binds into a pocket that’s not present in the unliganded native structure, a so-called “cryptic” binding site. Importantly, the binding site is not present in normal antibody heavy chain-light chain dimers, which means that the molecules should be specific for free light chains.
Coumarin 1 is not an ideal drug molecule. It doesn’t bind tightly enough to the light chain, and it will probably stick to a lot of other molecules in the body. We’re more excited about the binding site that we discovered. We will make molecules that bind much more tightly and specifically to light chains, based on the structure of the complex.
On the other hand, coumarin 1 is a fluorescent dye, and its fluorescence gets much brighter when it’s bound to light chains than when it’s free in solution. This means that we can use it as a probe for folded light chain dimers. This is not something that’s written on the bottle and it wouldn’t necessarily have been an obvious thing to try. But this “fluorogenicity” has been a big part of the lab’s research and it’s such a useful property that we routinely check for it in new compounds. In the paper, we use this property to ask how tightly the small molecule binds to different light chains, and whether other molecules bind in the same site. We hope that this will be a useful tool for other amyloidosis researchers.
I’ve spent three years working on this project and it’s great to see it published at last. It’s really the beginning of a process that we hope will lead to a drug for what is often an untreatable disease. There are a lot of challenges before we can think about clinical trials, but I’m confident that we will be able to make progress.