Research
Our research has focused on the development of novel agents that can elicit developmental stage-specific gene silencing or reprogramming in hematopoietic stem cells and progenitors through a series of epigenetic chromatin modifications by nucleosome remodeling, histone deacetylation, and demethylation. Such agents can be applied to the treatment of the patients with hematologic disorders such as sickle cell disease and beta-thalassemia, as well as acute myeloid leukemia (Blood cancer), which arise from congenital defects in the genome. The findings from these preclinical studies could also help to establish therapeutic indications in other hematological malignancies, and help to understand the basic mechanisms of hematologic development, function, and disease.
Ongoing Projects:
Discovery of a novel fetal hemoglobin (HbF) repressor complex, DRED
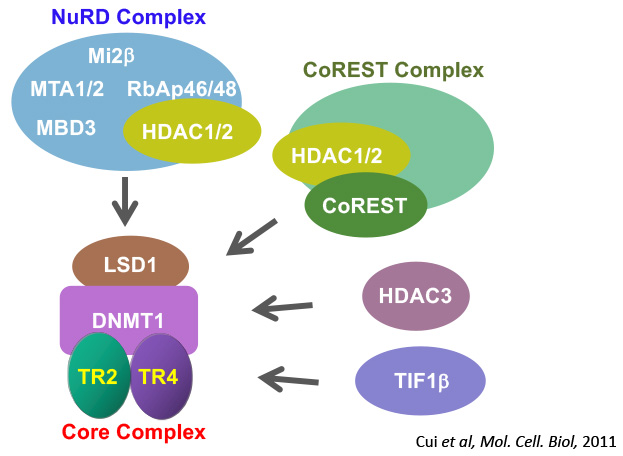
A decade ago, DRED (direct repeat erythroid definitive) was discovered in Dr. James Douglas Engel’s laboratory as a putative multi-protein transcriptional repressor complex of the human fetal globin genes. Our recent studies showed that DRED could assume multiple configurations depending on the associations that the TR2/TR4 heterodimer made with different co-repressor enzymes in the cellular milieu. The biochemical analyses of TR2/TR4 cofactors led to the identification of more than a dozen candidate co-repressor proteins that physically interact with TR2 and TR4. Those co-effector proteins (including DNMT1, NuRD, CoREST, LSD1, HDACs 1, 2 and 3, and TIF1β) are known to confer potent gene silencing through epigenetic chromatin modification. We further determined that a core complex, consisting of a TR2/TR4 heterodimer in association with LSD1 and DNMT1 [two epigenetic chromatin-modifying enzymes forms the basis for HbF repression]. Since small molecules that specifically target nuclear receptors comprise some 16% of the current drug market in the US, TR2 and TR4, as well as other co-repressors should be outstanding molecular targets for developing novel therapeutics for sickle cell disease and beta-thalassemia.
Developing pharmacological inhibitors of DRED components for HbF induction
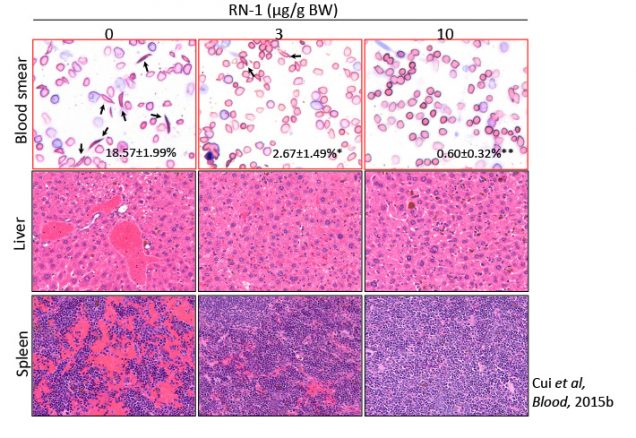
Our current efforts are focused on developing pharmacological inhibitors of DRED components, (especially TR2 and TR4, but also Dnmt1 and LSD1, which together form the tetrameric “core” complex of the repressor), as potential therapeutic targets. In particular, our research project aims to explore the potential of novel (and extremely powerful) HbF inducing agents based on LSD1 inhibition. LSD1 inhibitor RN-1 treatment of SCD mice resulted in increased γ-globin induction and HbF synthesis and led to the improvement of many aspects of disease pathology.
Discovery of a novel HbF co-activator, PGC-1a
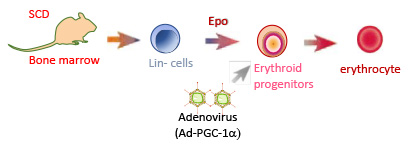
Our group recently identified PGC-1 alpha as a new protein involved in the regulation of the fetal globin genes. Forced overexpression of PGC-1 alpha in vitro by adenovirus infection in bone marrow cells from sickle cell disease mice significantly induced the expression of the gamma-globin genes. These data suggest that modulating PGC-1 alpha activity may be effectively applied to the treatment of sickle cell disease since enhanced HbF synthesis would alleviate the pathophysiological effects of sickle cell disease.
Targeting long non-coding RNA (LncRNA) in HbF regulation
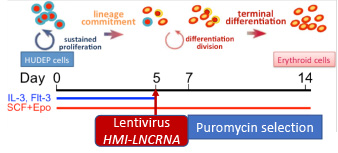
We recently characterized a novel erythroid lncRNA, which we named HMI-LNCRNA. Down-regulation of HMI-LNCRNA in the erythroid cell line (HUDEP-2) as well as in human primary hematopoietic progenitor CD34+ cells resulted in a significant increase of fetal γ-globin gene expression and HbF synthesis. Targeting this first LncRNA associated with HbF therapeutically has the advantages of increased specificity, different RNA-directed therapeutic approaches, and the possibility of small molecule inhibitors.