Research
Our research group is focused on computational materials science, with current emphasis on understanding excited-state phenomena within organic and ionic optoelectronic materials for renewable energy and display technologies. Much of this work utilizes first-principles density functional theory (DFT) and beyond DFT approaches that accurately predict measurable properties of materials. We often work closely with experimentalists in order to both validate our predictions and interpret measurements.
Current Projects
How does solid-state morphology affect optical excitations in organic materials?
Extended organic molecular assemblies are challenging for first-principles modeling due to long-range electron-electron and electron-phonon interactions. We apply DFT, TDDFT with long-range corrected exchange correlation functionals and GW/BSE to model these excitations, incorporating electron-phonon interactions by treating atoms as classical particles. Specifically, we are studying:
- Vibronic effects in organic photovoltaic materials
- Transport properties and electronic structure of conductive biopolymers
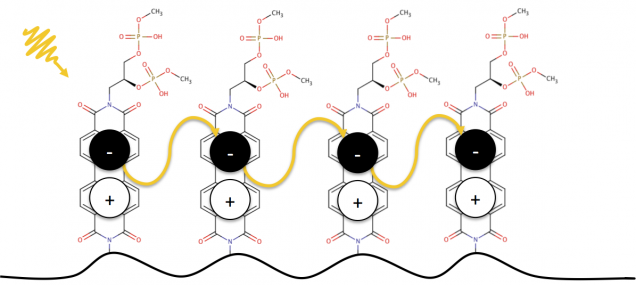
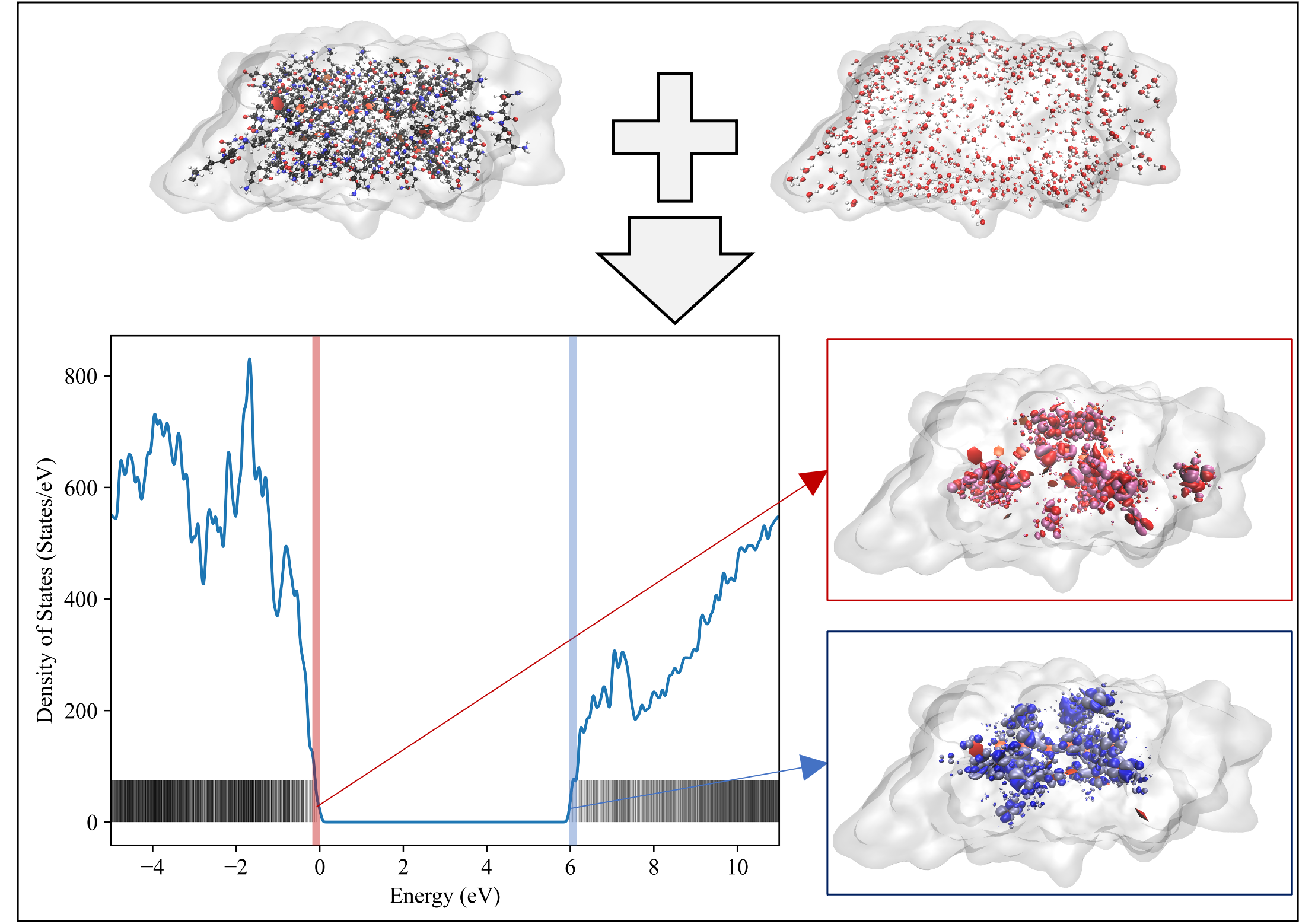
Electron-phonon interactions in low-dimensional and layered materials
Reduced dimensionality results in reduced solid-state screening and novel optoelectronic properties in materials. Understanding the interplay of electronic, nuclear, and collective excitations is critical in forming a robust, predictive understanding of their physical properties. For this class of materials, we are specifically interested in:
- The role of exciton-phonon interactions on the optical properties of 2D materials
- Predicting the Raman spectrum of metallophillically bound metal nanowires
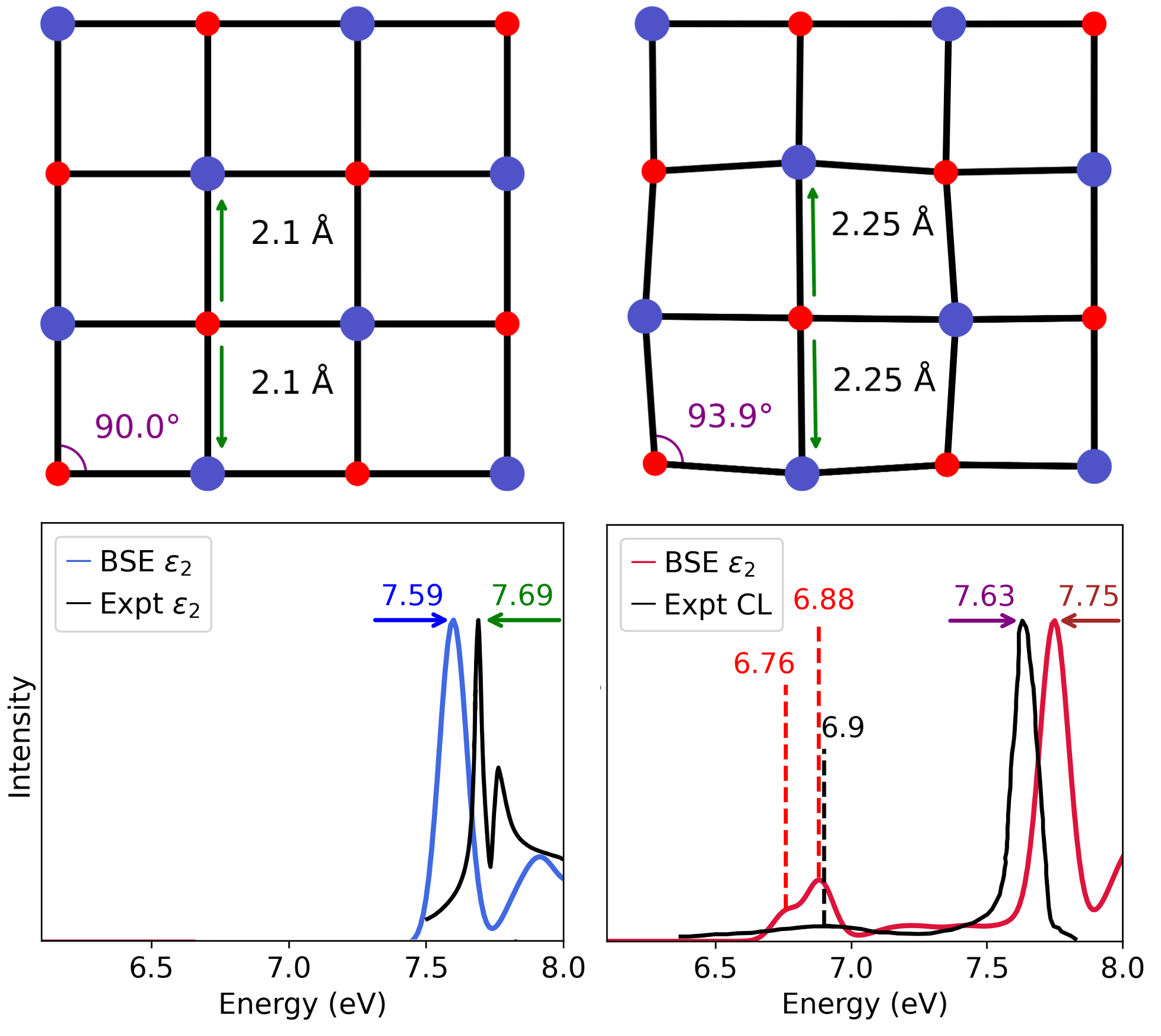
The Physics of Imperfections in Low-Dimensional Materials
Whether naturally occurring or synthesized, defects in materials can significantly influence optoelectronic properties. We aim to understand, and thus, control, the excited-state properties of defective materials. Specific projects of interest are:
- Localized point defects in 2D semiconductors
- Electronic structure of sp3 defects in single walled carbon nanotubes (SWCNTs) for quantum information science

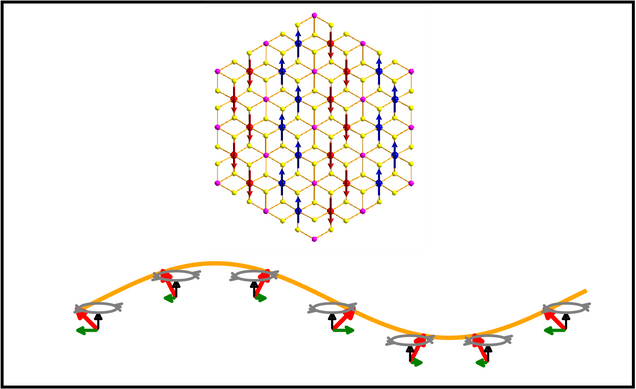
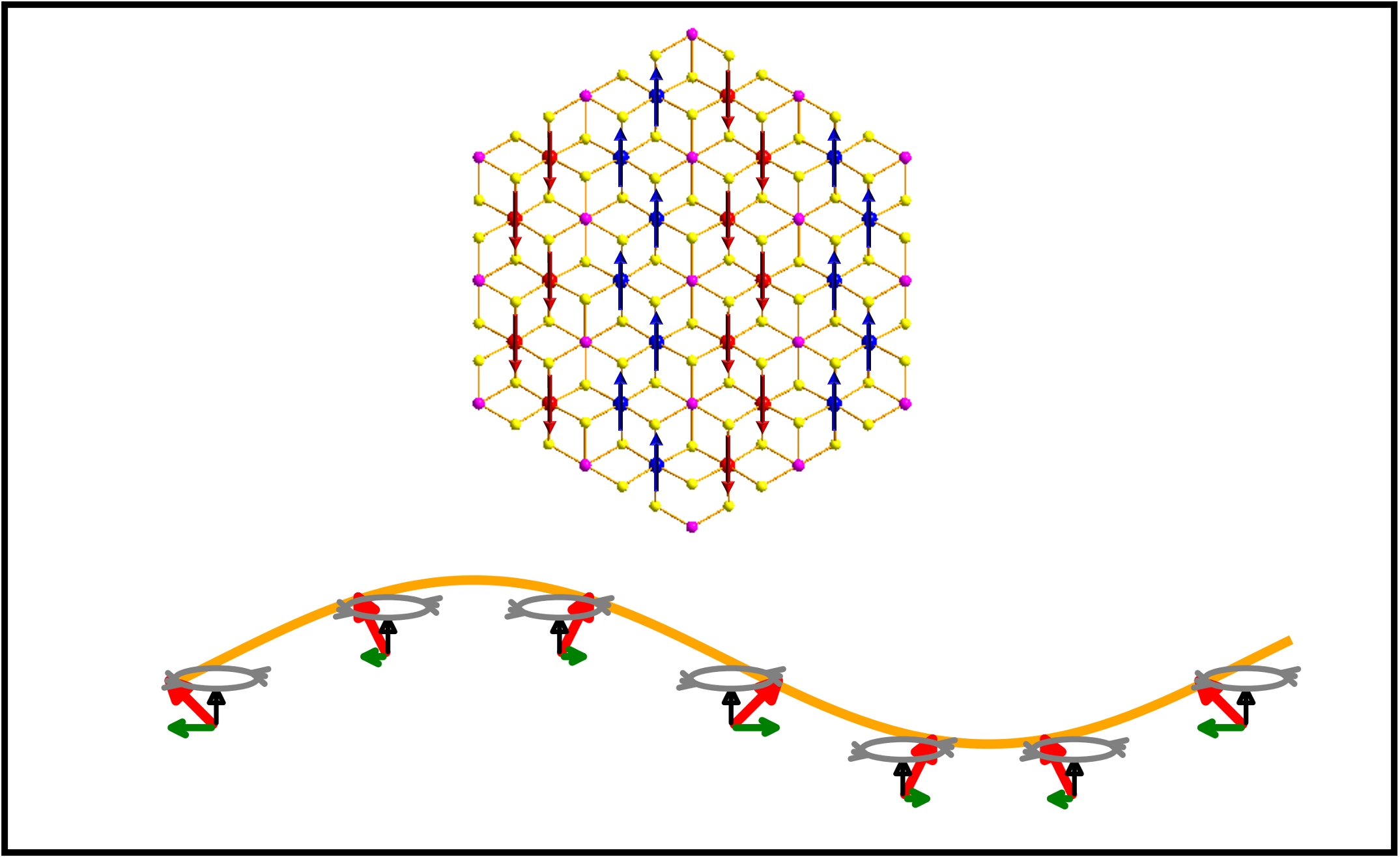
Correlated Spin–Excitonic Phenomena in Magnetic Materials
We are interested the interplay between spin, excitons, and lattice dynamics in layered and low-dimensional materials. Layered antiferromagnets such as NiPS₃ display striking excitonic features, including bright, narrow emission peaks and unusual magnetic-field responses, that arise from strong electron correlation and coupling between excitonic and spin degrees of freedom. Current projects include:
- Developing first-principles methodology to describe correlated exciton-spin physics
- Understanding optical properties of materials in the presence of spin fluctuations
Funding:
National Science Foundation
- DMR-1847774 “CAREER: First-Principles Investigation of Energy Transport Within Ordered Organic Assemblies” (2019-2024)
Department of Energy
- DE-SC0026296 “Moiré Chemistry Toward the Synthesis of Spatially Selective Functionalization of 2D Materials” (2025-2028)
Army Research Office
- W911NF2510245 “Understanding Spin Correlated Excitons in Magnetic Materials from First-Principles Theory” (2025-2026)
![]()