New paper: Structural Basis for the Stabilization of Amyloidogenic Immunoglobulin Light Chains by Hydantoins
How does a curved molecule fit into a flat binding pocket?
Structural basis for the stabilization of amyloidogenic immunoglobulin light chains by hydantoins.
Nicholas L. Yan, Diogo Santos-Martins, Enrico Rennella, Brittany B. Sanchez, Jason S. Chen, Lewis E. Kay, Ian A. Wilson, Gareth J. Morgan, Stefano Forli, Jeffery W. Kelly
Bioorganic & Medicinal Chemistry Letters
Volume 30, Issue 16, 15 August 2020, 127356
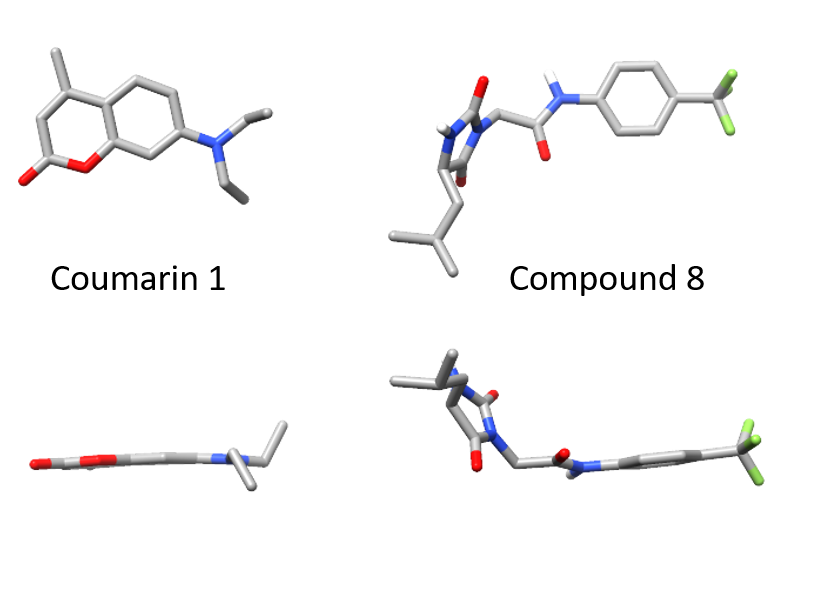
When we identified small molecules that can stabilize antibody light chains, aiming to develop a drug for AL amyloidosis, most of the hits from our screen were flat molecules. The molecule that we investigated most thoroughly, coumarin 1, has a two-ring aromatic core that slides in between two domains of the light chain dimer. The overall binding pocket, which is not present in the unbound light chain structure, is deep and flat. While this allows for reasonably tight binding, flat, hydrophobic molecules are generally not ideal as drug candidates because they tend to bind to many different proteins. Coumarins are generally regarded as “promiscuous” in this sense. We therefore had a potentially long road ahead in order to design a stabilizer molecule that binds tightly and specifically to light chains. Our new paper describes our first step on that road.
Of the twenty or so molecules that came out of our initial screen, two related molecules are much less flat than the others. Both contain a 5-membered hydantoin ring at their core, linked to a trifluoromethylated phenyl ring. We called them coumponds 7 and 8. It was not obvious how these molecules could bind to the light chain, even though we had competition binding data that was consistent with them binding at the same site as the coumarins.
I’d previously met Stefano Forli at Scripps happy hour and we’d talked about doing some computational modeling of potential stabilizer molecules. (Go meet your colleagues, folks!) Stefano works on computational drug design and in silico modeling. Stefano’s postdoc, Diogo Santos-Martins, was able to carry out computational docking experiments that suggested a potential binding conformation of the hydantoin molecules. The phenyl ring and its trichloromethyl group packs into the known coumarin binding site, but the hydantoin and the rest of the molecule stick out, interacting with new residues on the surface of the light chain.
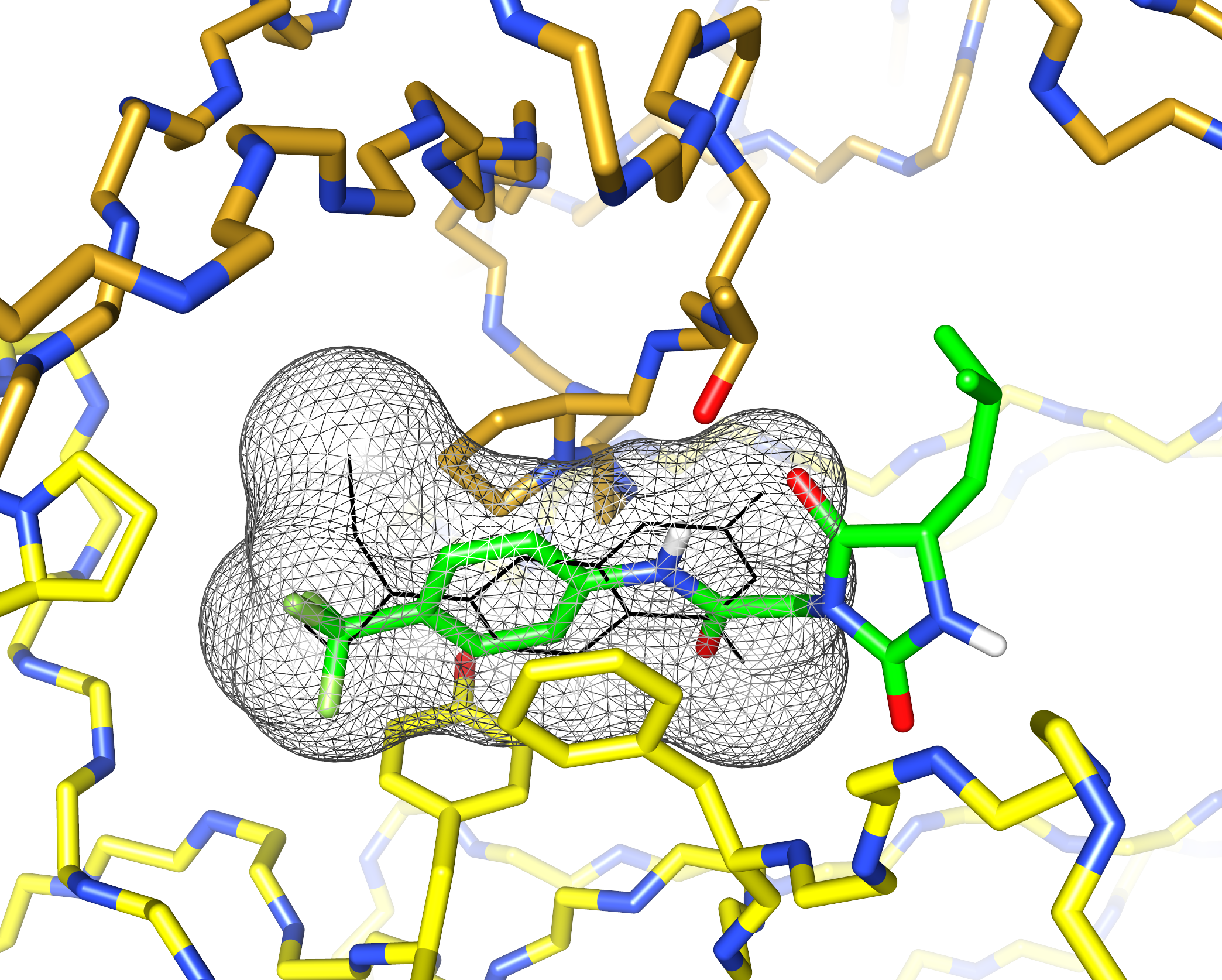
This was exciting because increasing the number of interactions between a protein and its ligand is a good way to improve affinity and specificity. Nick Yan, a graduate student working with me in the Kelly lab at Scripps, set about making derivatives of these molecules and managed to crystalize one of the complexes. The X-ray structure showed almost exactly the same binding conformation as the computational model. We verified that this also happens in solution, using NMR and analytical ultracentrifugation.
One difference was in the stereochemistry of the structure. Nick used a racemic mixture of the two forms of the hydantoin molecule in his experiments, but only one form was visible in the crystal. Diogo showed that this enantiomer bound more tightly in the simulations. Nick separated the two forms and found that one was able to protect light chains from proteolysis more effectively than the other.
This paper is a neat demonstration of how computational tools can help to prioritize screening hits. The new interactions that Diogo’s work pointed out have allowed Nick to design and synthesize more effective small molecule stabilizers. That work is still in progress, but we’re moving in the right direction.
This link has free access to the full paper until August 2020, but might need you to suspend your pop-up blocker: https://authors.elsevier.com/c/1bIAs,LsIFmv4Y