
Image Summaries
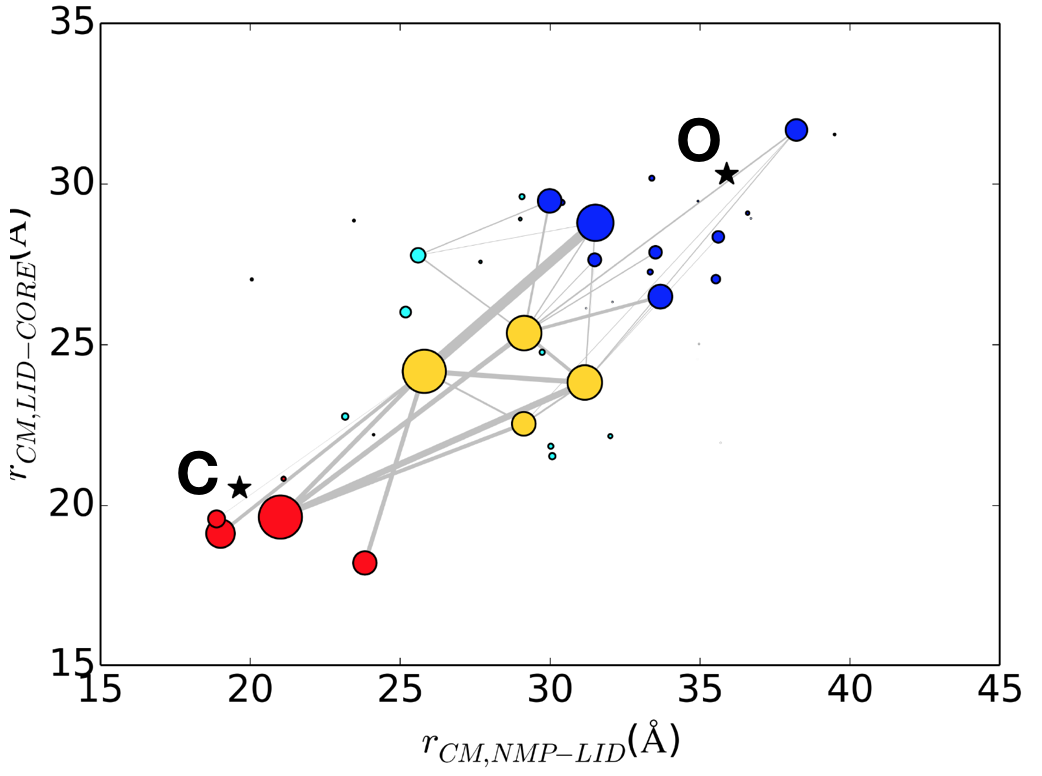
Multiple pathways and time scales of protein transitions: Using extensive (~50 microsec) explicit solvent
atomistic simulations and Markov state analysis, we shed new lights on the mechanism of open/close transition in the apo form of AK. Our results highlighted the existence of multiple pathways and potentially multiple time scales for such functional transitions. Besides interdomain interactions, a novel mutual information analysis identifies specific intradomain interactions that correlate strongly to transition kinetics. See J. Chem. Theory Comp. (2018)
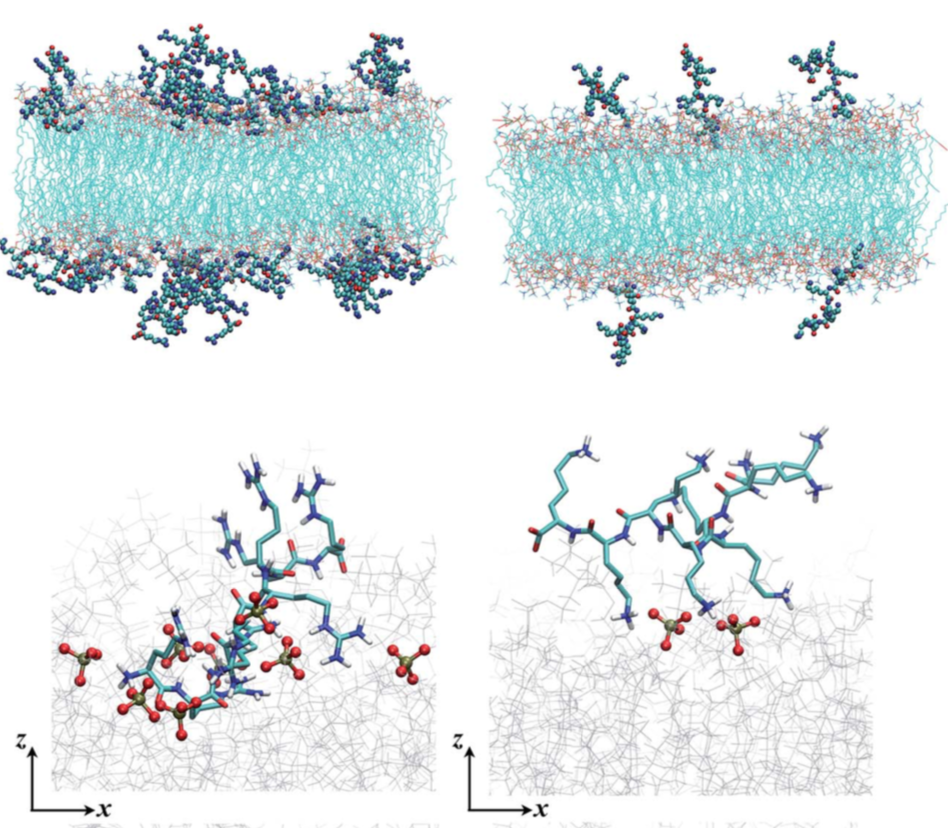
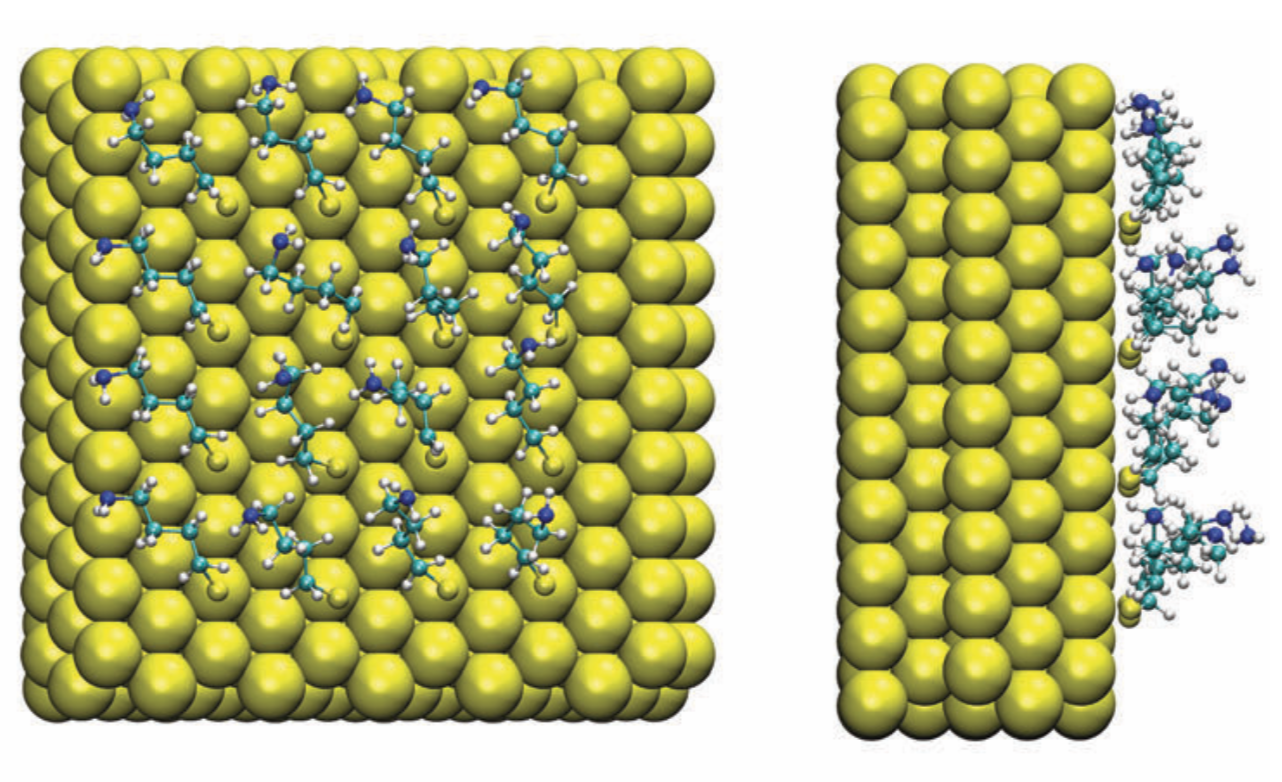
Poly-Arg vs. Poly-Lys at membrane surface: Quantifying the number of charges on peptides bound to interfaces requires reliable estimates of (i) surface coverage and (ii) surface charge, both of which are notoriously difficult parameters to obtain, especially at solid/water interfaces. By combining non-linear spectroscopies (SHG), mass sensing measurements (QCMD and NPS) and atomistic molecular dynamics simulations, we were able to quantify the amount of charge and adsorption for poly-Arg/poly-Lys on membrane surfaces. Analysis of electrostatic potential and charge distribution from atomistic simulations suggests that the Gouy–Chapman model, which is widely used for mapping surface potential to surface charge, is semi-quantitatively valid. See Chem. Sci. (2018)
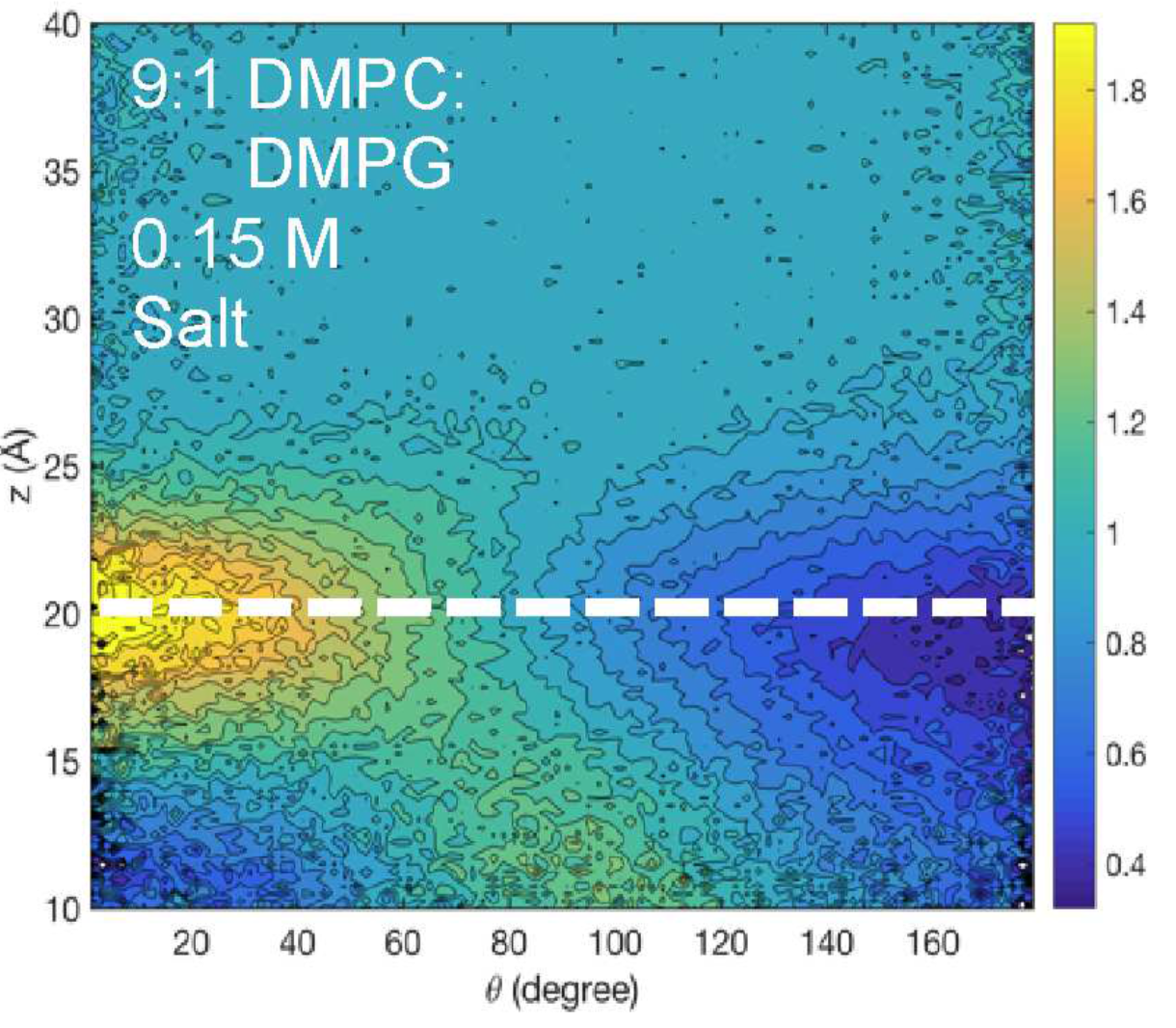
Behavior of water at lipid membrane interface: By combining sum-frequency generation (SFG) spectroscopy and atomistic simulations, we find that the strength and orientation distribution of the hydrogen bonds over the purely zwitterionic bilayers are largely invariant between submicromolar and hundreds of millimolar concentrations. However, specific interactions between water molecules and lipid headgroups are observed upon replacing phosphocholine (PC) lipids with negatively charged phosphoglycerol (PG) lipids, which coincides with SFG signal intensity reductions in the 3100−3200 cm−1 frequency region. The atomistic simulations show that this outcome is consistent with a small, albeit statistically significant, decrease in the number of water molecules adjacent to both the lipid phosphate and choline moieties per unit area, supporting the SFG observations. See J. Phys. Chem. B (2018)
Multi-scale analysis of solid/liquid interface: We describe a strategy of integrating quantum mechanical (QM), hybrid quantum mechanical/molecular mechanical (QM/MM) and MM simulations to analyze the physical properties of a solid/water interface. This protocol involves using a correlated ab initio (CCSD(T)) method to first calibrate Density Functional Theory (DFT) as the QM approach, which is then used in QM/MM simulations to compute relevant free energy quantities at the solid/water interface using a mean-field approximation of Yang et al. that decouples QM and MM thermal fluctuations; gas-phase QM/MM and periodic DFT calculations are used to determine the proper QM size in the QM/MM simulations. Finally, the QM/MM free energy results are compared with those obtained from MM simulations to directly calibrate the force field model for the solid/water interface. The strategy of integrating multiple computational methods to cross-validate each other for complex interfaces is applicable to many problems that involve both inorganic/metallic and organic/biomolecular components, such as functionalized nanoparticles. See Phys. Chem. Chem. Phys. (2018)
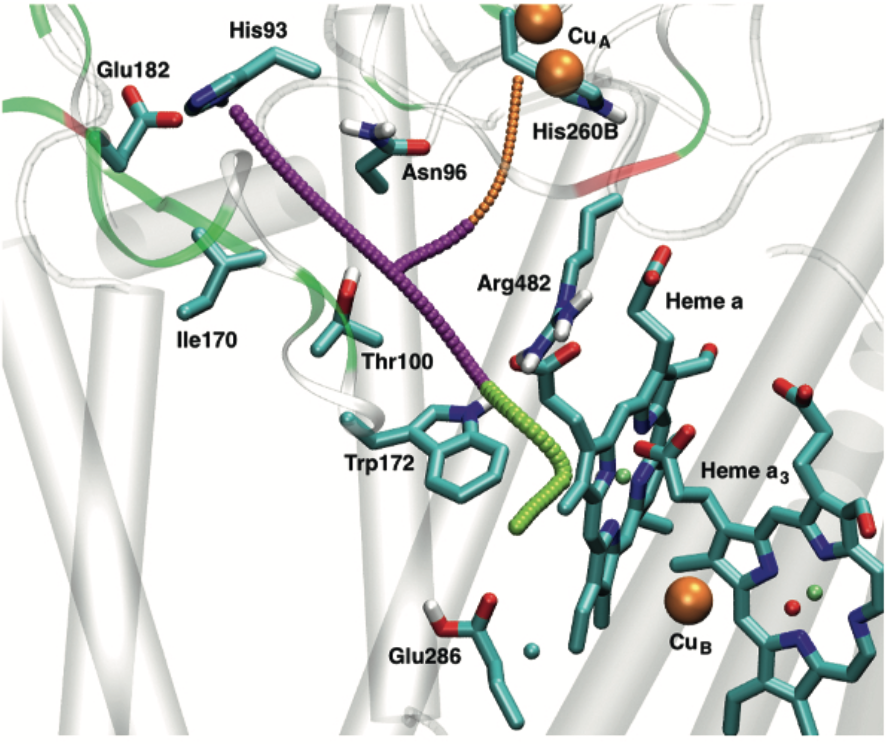
Reversible wetting/dewetting of a protein cavity: Cytochrome c oxidase (CcO) reduces molecular oxygen to generate the proton motive force across the membrane that drives ATP synthesis. Internal water molecules in and near a central cavity play important roles in mediating the proton transfers. Molecular simulations of CcO reveal reversible transitions between wet and dry configurations of this internal cavity in response to the charge state of key cofactors and residues. Quantitative analysis of the free energy change and timescale of the transition suggests that hydration-level change of the central cavity is an essential feature that con- tributes to the vectorial efficiency of proton pumping in CcO. Thus, wetting transition of protein internal cavities can be functionally significant, especially for the transport of charged species. See PNAS (2018)
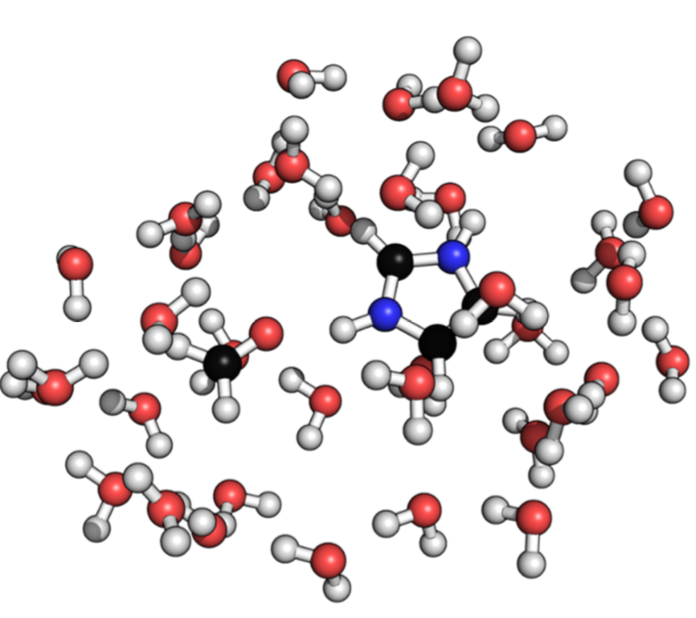
Analysis of Intermolecular interactions in water: To facilitate further development of approximate quantum mechanical methods for condensed phase applications, we present a new benchmark dataset of intermolecular interaction energies in the solu- tion phase for a set of 15 dimers, each containing one charged monomer. The reference interaction energy in solution is computed via a thermodynamic cycle that integrates dimer binding energy in the gas phase at the coupled cluster level and solute-solvent interaction with density functional theory. As most approximate QM methods are parametrized and evaluated using data measured or calculated in the gas phase, the dataset represents an important first step toward calibrating QM based methods for application in the condensed phase. See J. Chem. Phys. (2017)

Benchmark of DFTB for organic reaction transition states: To establish the applicability of DFTB models to general chemical reactions, we conduct benchmark calculations for barrier heights and reaction energetics of organic molecules using existing databases and several new ones compiled in this study. Structures for the transition states and stable species have been fully optimized at the DFTB level, making it possible to characterize the reliability of DFTB models in a more thorough fashion. The encouraging results for the diverse sets of reactions studied here suggest that DFTB models, espe- cially the most recent third-order version (DFTB3/3OB aug- mented with dispersion correction), in most cases provide satisfactory description of organic chemical reactions with accuracy almost comparable to popular DFT methods with large basis sets, although larger errors are also seen for certain cases. See J. Comput. Chem. (2017)
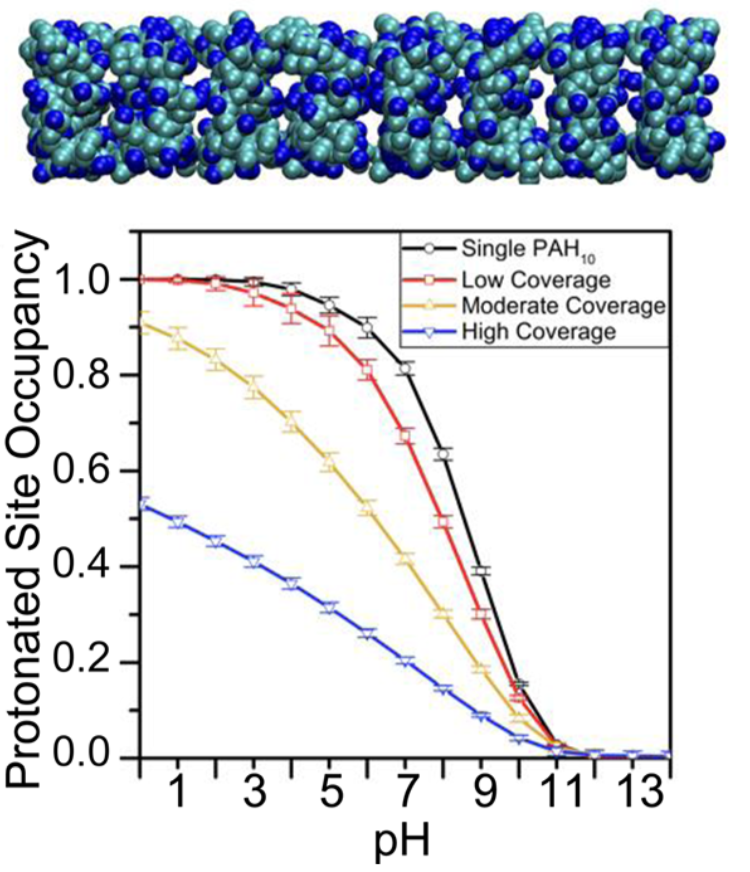
Adsorption of polyelectrolyte at membrane surface: Mechanistic insight into how polycations disrupt and cross cell membranes is needed for understanding and controlling polycation−membrane interactions, yet such information is surprisingly difficult to obtain at the molecular level. We use second harmonic and vibrational sum frequency generation spectroscopy along with quartz crystal microbalance with dissipation monitoring and computer simulations to quantify the interaction of poly(allylamine) hydrochloride (PAH) and its monomeric precursor allylamine hydrochloride (AH) with lipid bilayers. By experimentally quantifying the number of adsorbates and the average amount of charge carried by each adsorbate, we find that the PAH is associated with only 70% of the positive charges it could hold. Our results provide experimental constraints for theoretical calculations, which yield atomistic views of the structures that are formed when polycations interact with lipid membranes that will be important for predicting polycation-membrane interactions. See JACS. (2017)
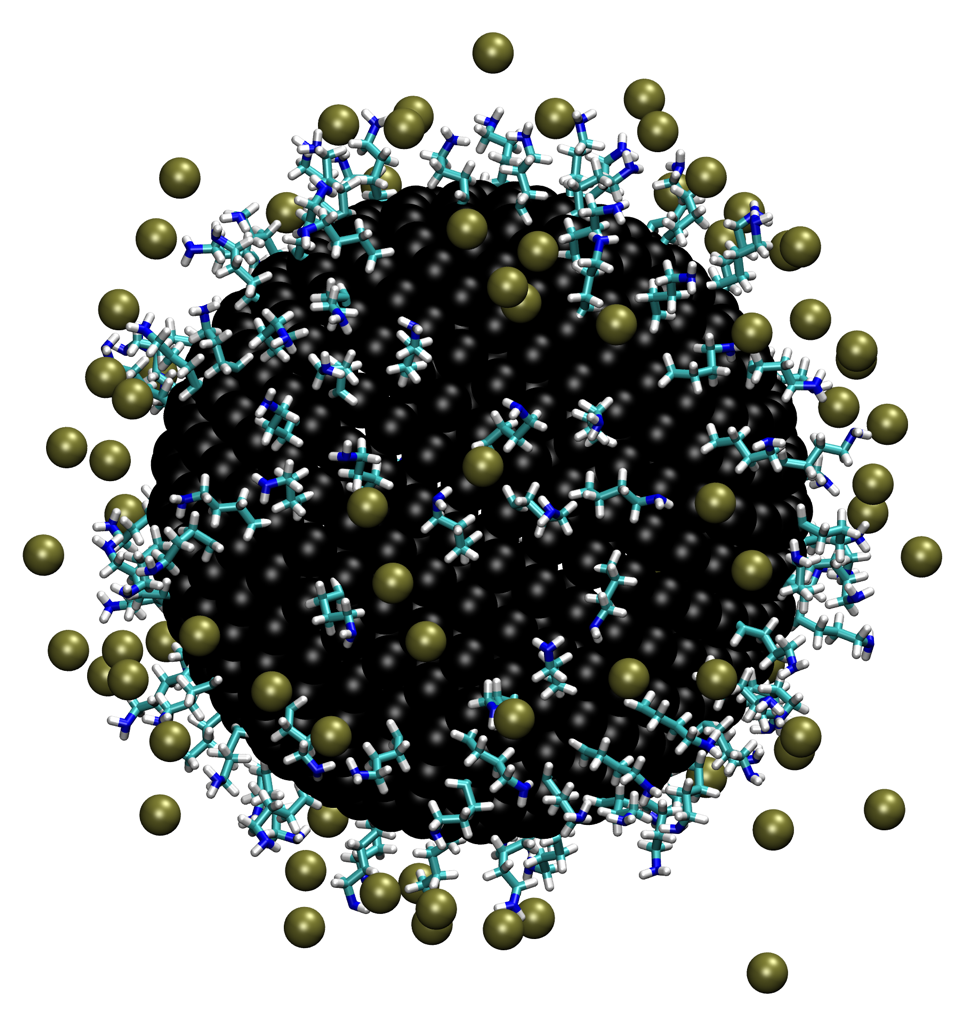
Determination of charge of functionalized nanoparticle: The surface charge of nanomaterials determines their stability in solution and interaction with other molecules and surfaces, yet experimental determination of surface charge of complex nanomaterials is not straightforward. We propose a hybrid approach that iteratively integrates explicit solvent molecular dynamics simulations and a multiconformer continuum electrostatic model (MCCE) to efficiently sample the configurational and titration spaces of surface ligands of nanomaterials. Test calculations of model systems indicate that the iterative approach converges rapidly even for systems that contain hundreds of titratable sites, making the approach complementary to more elaborate methods such as explicit solvent-based constant-pH molecular dynamics. The hybrid computational approach makes a major step toward guiding the design of nanomaterials with desired charge properties. See J. Phys. Chem. C (2017)
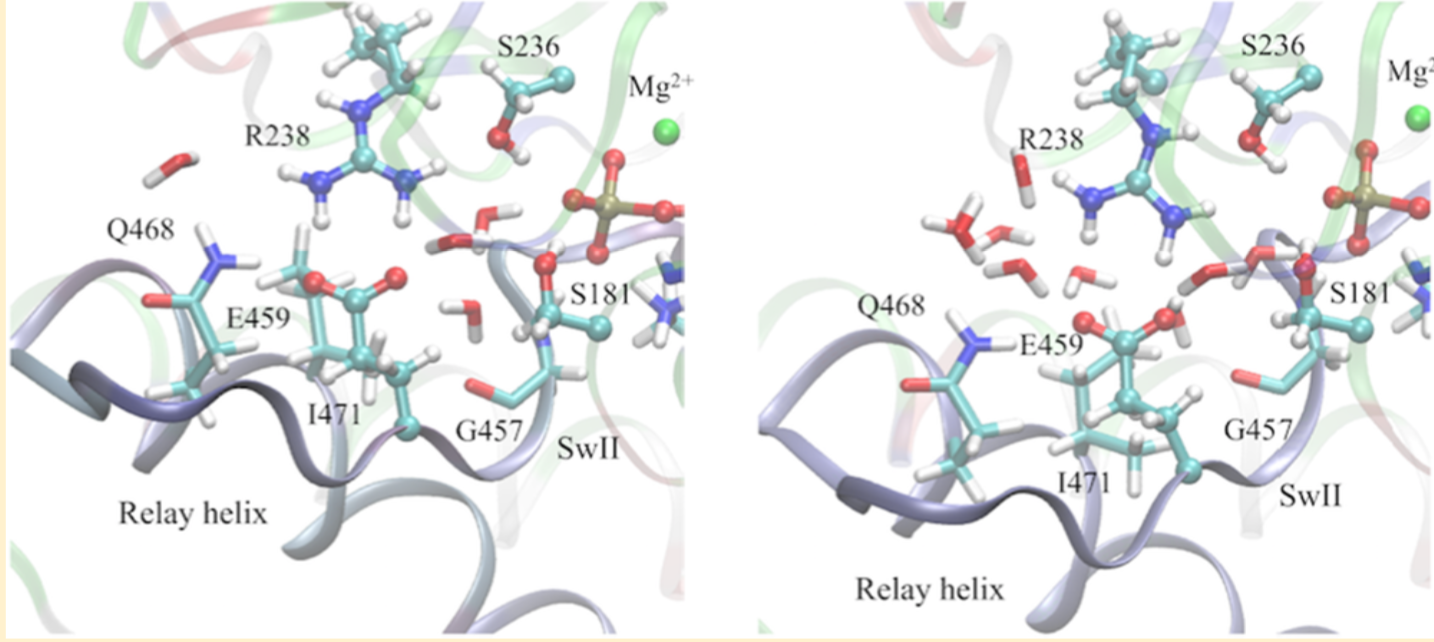
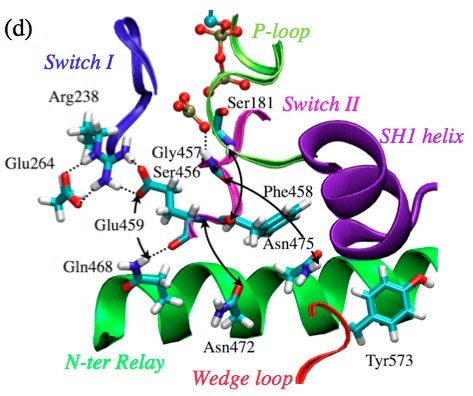
Regulation of ATP hydrolysis in myosin: With extensive DFTB3/MM free energy simulations, the mechanism of ATP hydrolysis in the myosin motor domain is analyzed in detail. The ATP hydrolysis activity is found to depend on the positioning of nearby water molecules, and a network of polar residues facilitates proton transfer and charge redistribution during hydrolysis. Proton transfer from the lytic water to the γ-phosphate through active site residues is an important part of the kinetic bottleneck; several hydrolysis pathways that feature distinct proton transfer routes are found to have similar free energy barriers, suggesting a significant degree of plasticity in the hydrolysis mechanism. Comparison of hydrolysis in the pre-powerstroke state and the closed post-rigor model suggests that optimization of residues beyond the active site for electrostatic stabilization and preorganization is likely important to enzyme design. See Biochem. (2017)
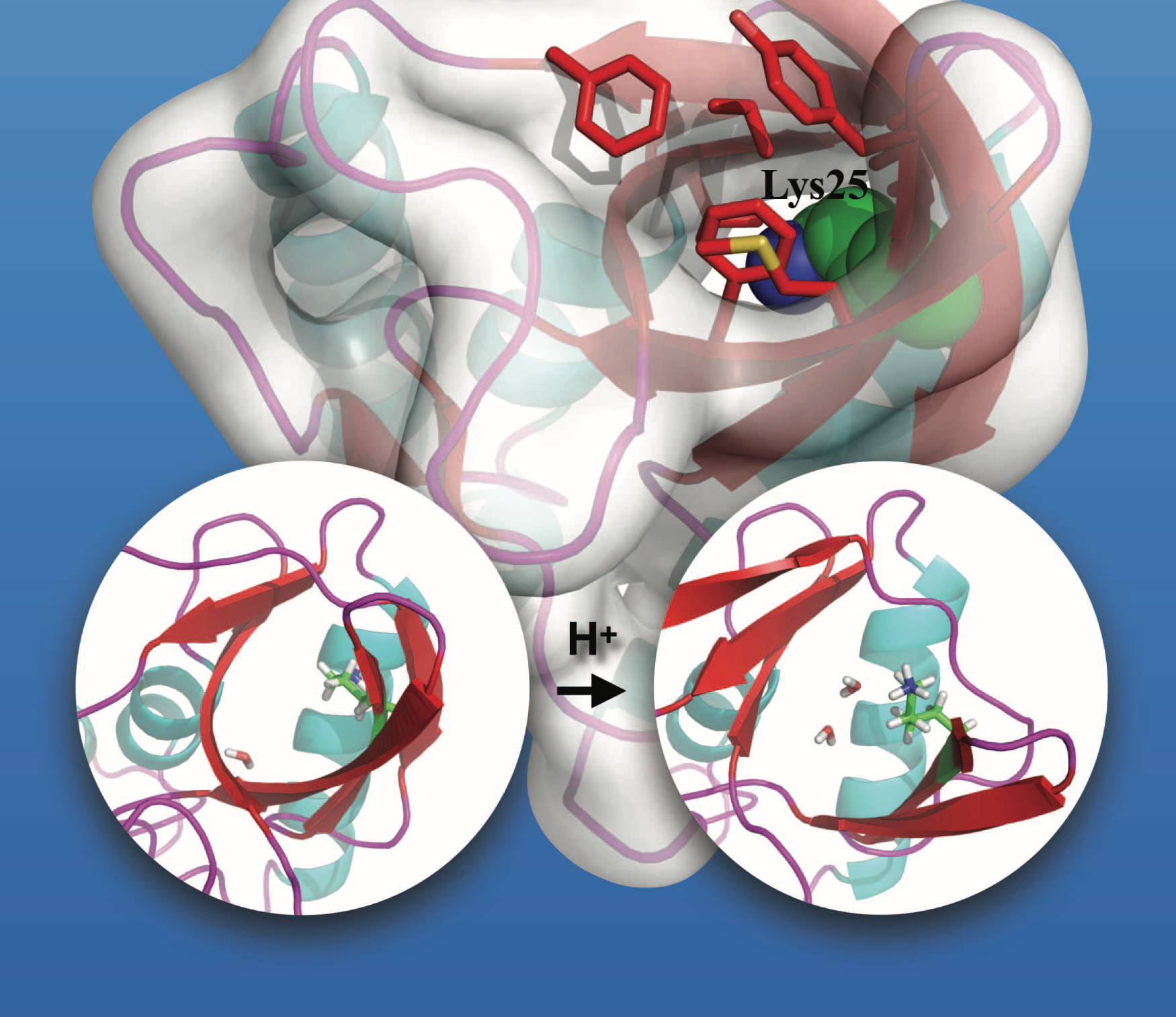
Microscopic Behavior for the titration of a buried residue:
To probe the microscopic mechanisms that govern the titration behavior of buried ionizable groups, microsecond explicit solvent molecular dynamics simulations are carried out for several mutants of Staphylococcal nuclease using both fixed charge and polarizable force fields. Overall, the set of unbiased simulations provides insights into the spatial and temporal scales of protein and solvent motions that dictate the diverse titration behaviors of buried protein residues. Using the sampled conformational ensembles, good agreement with experimental pKa values is obtained with Poisson-Boltzmann calculations using a protein dielectric constant of 2-4 for V66D/E; slightly larger dielectric constants are needed for Lys mutants especially L25K, suggesting that structural responses beyond microseconds are involved in ionization of Lys 25. Proteins , 85, 268-281 (2017)
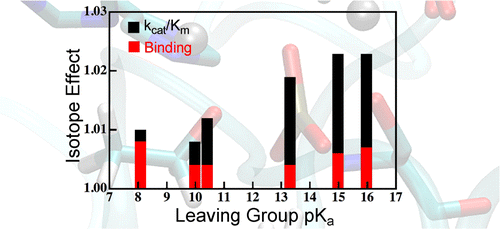
Interpretation of KIE in metalloenzyme:
We have used quantum mechanics calculations and hybrid quantum mechanics/molecular mechanics (QM/MM) simulations to model a variety of isotope effects relevant to the reaction of AP. We have calculated equilibrium isotope effects (EIEs), binding isotope effects (BIEs), and kinetic isotope effects (KIEs) for a range of phosphate mono- and diester substrates. The results agree well with experimental values, but the model for the reaction’s transition state (TS) differs from the original interpretation of those experiments. Our model indicates that isotope effects on binding make important contributions to measured KIEs on V/K, which complicated interpretation of the measured values. J. Am. Chem. Soc. , 138, 11946-11957 (2016)
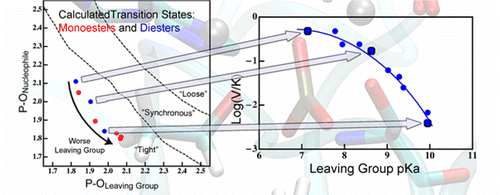
Nature of Transition State in metalloenzyme:
A reaction’s transition state (TS) structure plays a critical role in determining reactivity and has important implications for the design of catalysts, drugs, and other applications. In the enzyme alkaline phosphatase, using hybrid Quantum Mechanics/Molecular Mechanics simulations, we find that minor perturbations to the substrate have major effects on TS structure and the way the enzyme stabilizes the TS. The results predict nonlinear free energy relationships for a single rate-determining step, and substantial differences in kinetic isotope effects for different substrates; both trends were observed in previous experimental studies, although the original interpretations differed from the present model. Our results demonstrate the considerable plasticity of TS structure and stabilization in enzymes. Furthermore, perturbations to reactivity that probe TS structure experimentally (i.e., substituent effects) may substantially perturb the TS they aim to probe, and thus classical experimental approaches such as free energy relations should be interpreted with care. J. Am. Chem. Soc. , 138, 7386-7394 (2016)
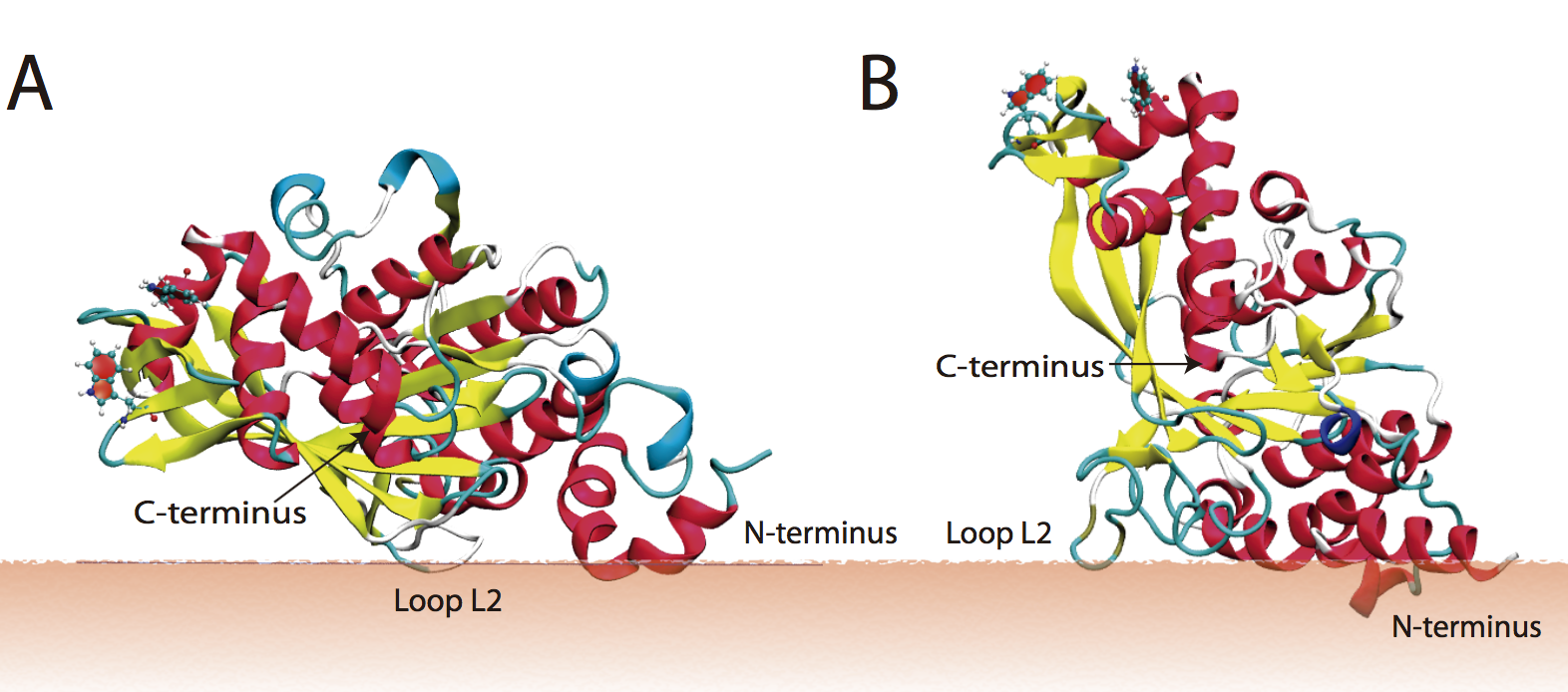
Binding properties of peripheral protein and lipids:
We describe a computational and experimental approach for probing the binding properties of the RecA protein at the surface of anionic membranes. Fluorescence measurements indicate that RecA behaves differently when bound to phosphatidylglycerol (PG)- and cardiolipin (CL)-containing liposomes. We use a multistage computational protocol that integrates an implicit membrane/solvent model, the highly mobile mimetic membrane model, and the full atomistic membrane model to study how different anionic lipids perturb RecA binding to the membrane. Overall, we find that ionic hydrogen bonds and lipid packing defects collectively determine the binding orientation and insertion depth of RecA on multicomponent lipid bilayers. J. Phys. Chem. B , 120, 8424-8437 (2016)
DFTB and VALBOND for Cu:
We apply two recently developed computational methods, DFTB3 and VALBOND, to study copper oxidation/reduction processes in solution and protein. The properties of interest include the coordination structure of copper in different oxidation states in water or in a protein (plastocyanin) active site, the reduction potential of the copper ion in different environments, and the environmental response to copper oxidation. Overall, the study supports the value of VALBOND and DFTB3(/MM) for the analysis of fundamental copper redox chemistry in water and protein, and the results also help highlight areas where further improvements in these methods are desirable. J. Phys. Chem. B , 120, 1894-1910 (2016)
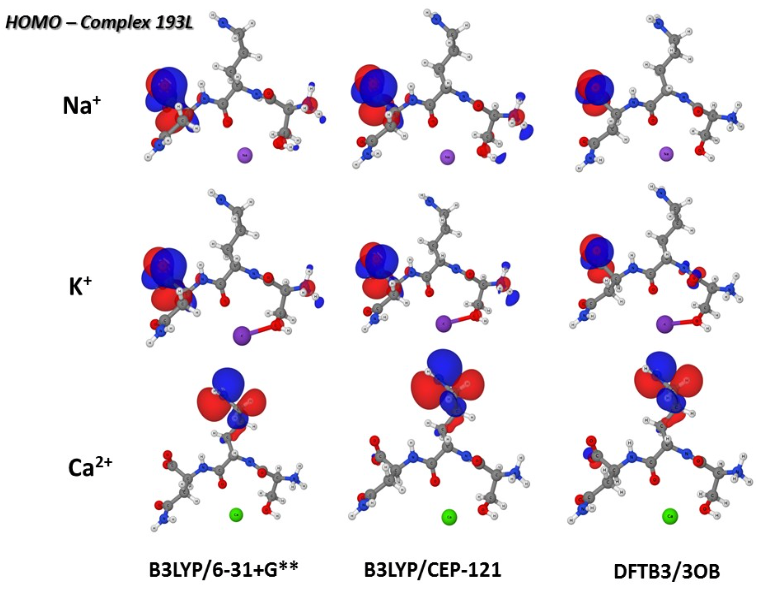
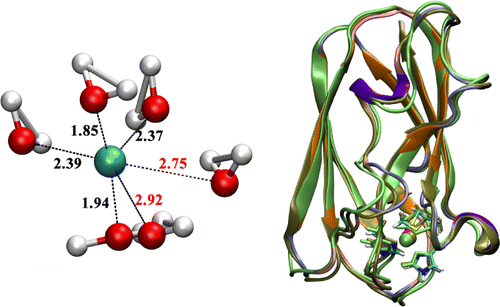
Polarization in metal coordination:
Metal ion coordination is essential to the structure and function of metalloproteins. It is thus important to understand the value and limitation of various computational approaches for describing metal ions in proteins. In this collaborative study, we compare non-polarizable and polarizable molecular mechanics force fields, the DFTB3 model and DFT methods for the description of Na+, K+ and Ca2+ binding to proteins. The results highlight the importance of polarization and charge transfer to metal-protein interactions, especially for the divalent calcium ion. J. Chem. Theo. Comp. , 11, 4992-5001 (2015)
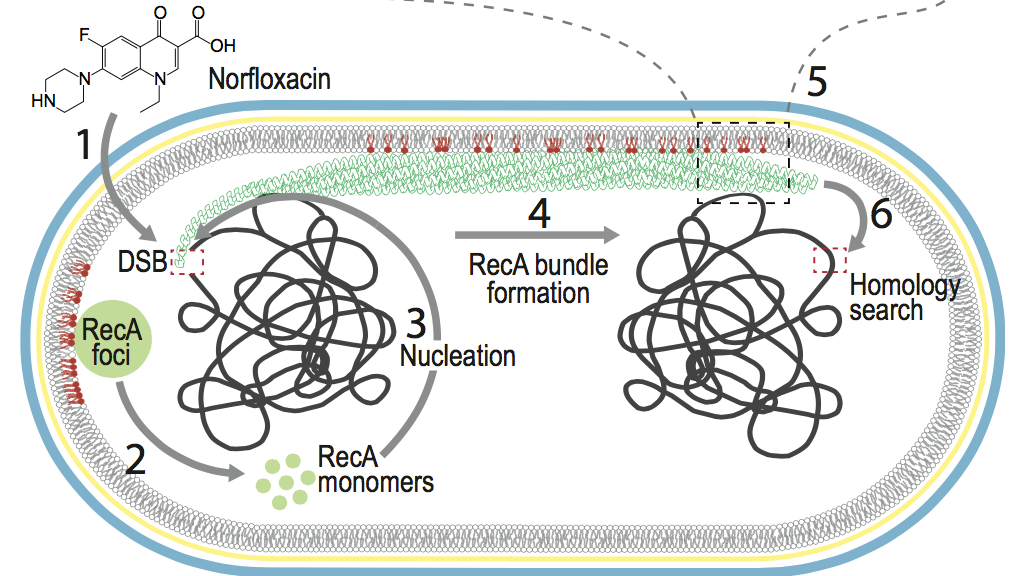
Cellular localization of RecA by membrane:
Factors that regulate the cellular localization of proteins often remain unclear. In this combined experimental and computational study, we show that anionic lipids (PG and cardiolipin) play an essential role in localizing RecA to bacterial cell poles. Binding to the membrane modifies the ATP hydrolysis activity of RecA and therefore stabilizes RecA filaments, which are critical to the initiation of the SOS response in bacteria. Mol. Cell , 60, 374-384 (2015)
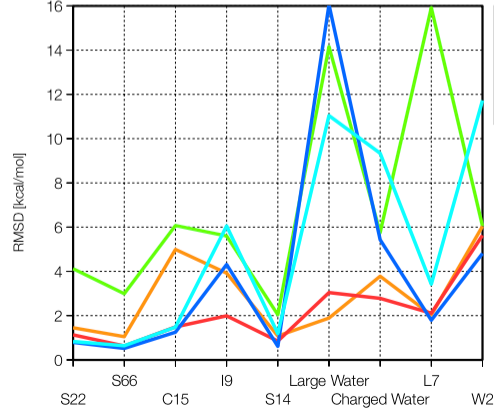
Polarization in DFTB3:
Being a minimal basis approach, DFTB3 underestimates electronic polarization and therefore is less accurate for the description of non-covalent interactions that involve highly polar/charged motifs. By including an auxiliary set of basis functions that describes the electronic response properties and solving the expansion coefficients in the framework of chemical potential equilization, we are able to substantially improve this limitation of DFTB3. By further including an empirical dispersion (D3) model, we show that DFTB3/CPE-D3 offers a balanced treatment of a broad range of non-covalent interactions. J. Chem. Phys. , 143, 084123 (2015)
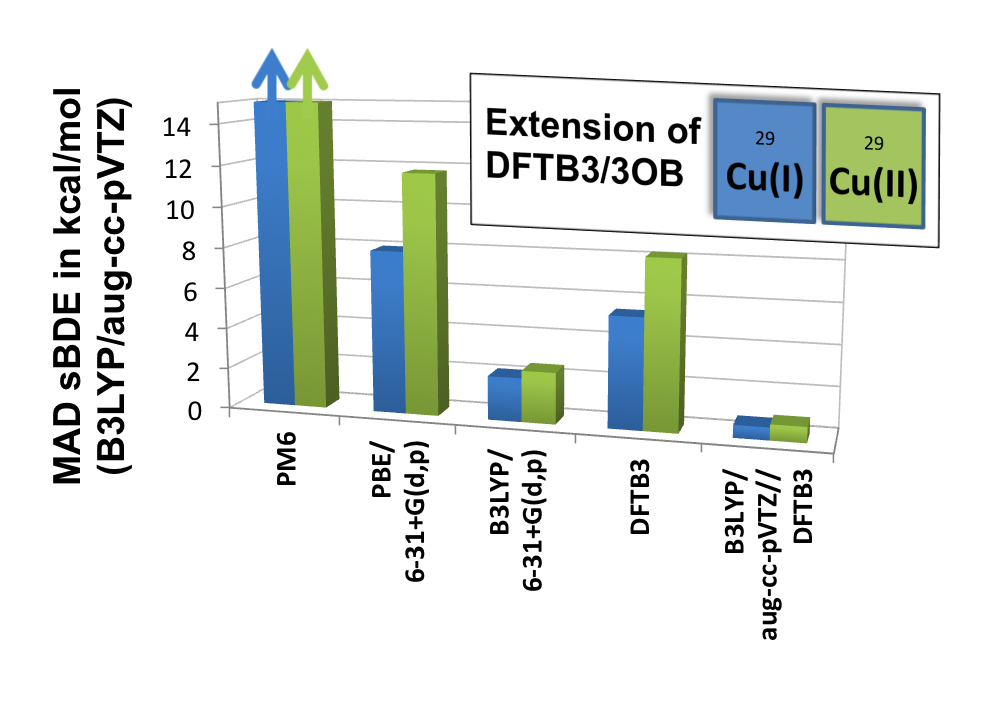
DFTB3/3OB for Copper:
Transition metal ions are essential to the structure and function of many biomolecules. To facilitate computational analysis of these systems, esepcially those are structurally flexible, we have initiated efforts in parameterizing the DFTB3 model for transition metal ions, for which traditional semi-empirical models have had only limited success. We show that by including the orbital angular momentum dependence of the Hubbard parameter and its charge derivative in the DFTB3 framework, we are able to provide a balanced description of both oxidation states of copper, especially for structural properties. J. Chem. Theo. Comp. , 11, 4205-4219 (2015)
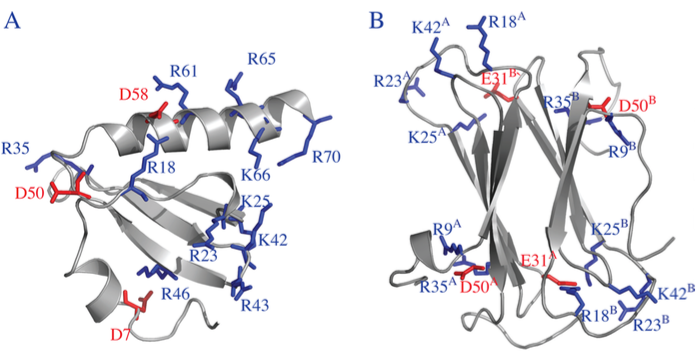
Metamorphic protein hLtn:
Human lymphotactin (hLtn) is a remarkable example of a metamorphic protein; i.e., it completely changes fold upon variation of the solution condition. Using molecular dynamics simulation, we gain new insights into the interplay of electrostatic and hydrophobic interactions that modulate the relative stability of the two folds. The analysis leads to predictions of functionally important residue pairs that can be tested experimentally. J. Phys. Chem. B , 119, 9547-9558 (2015)
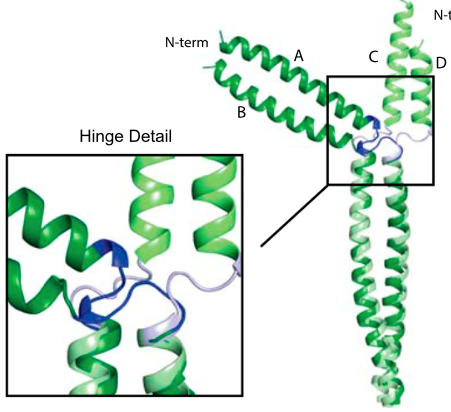
Skip residues in myosin rod assembly:
Myosin rods consist of filaments that are largely coiled-coils (CCs) but interrupted by several conserved “skip residues” that break the regular CC pattern. By combining crystallography, molecular dynamics simulations and in vivo imaging studies, we establish the physiological roles of these skip residues. In particular, we show that the fourth skip residue behaves very differently from the other three and is essential for the assembly of the rods. PNAS, E3806 (2015)
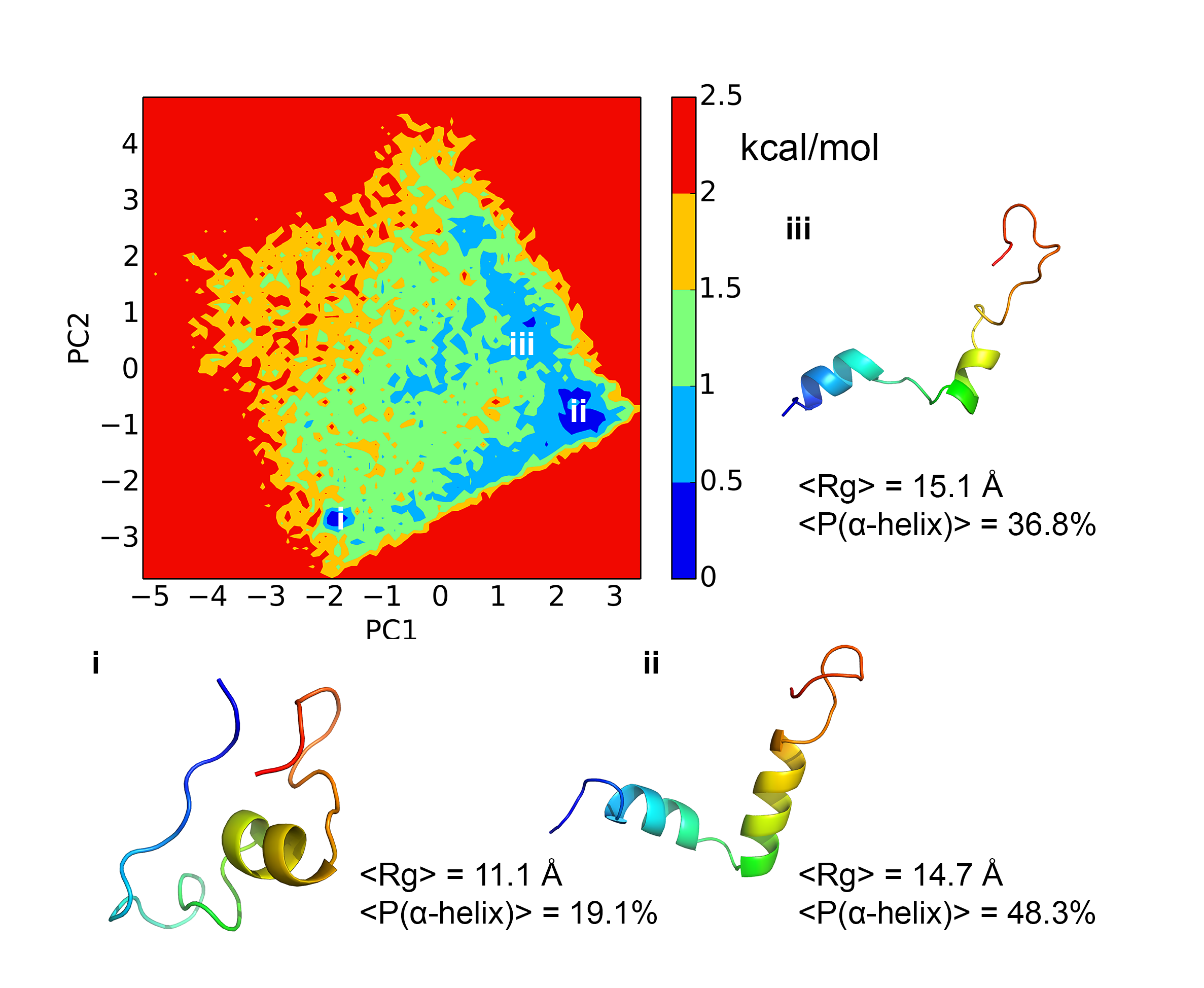
Free energy landscape and kinetics of H3 tail:
Histone tails play a critical role in regulating chromatin dynamics and gene activity. We have carried out extensive explicit solvent simulations for the H3 tail to characterize not only the underlying free energy landscape but also kinetic properties through Markov state models. Dihedral principal component analysis and locally scaled diffusion map analysis yield consistent results that indicate an overall flat free energy surface with shallow basins that correspond to conformations with a high alpha-helical propensity in two regions of the peptide. Kinetic information extracted from Markov state models reveals rapid transitions between different metastable states with mean first passage times spanning from several hundreds of nanoseconds to hundreds of microseconds. These findings shed light on how the dynamical nature of histone H3 N-terminal tail is related to its function. PCCP, 17, 13689-13698 (2015)
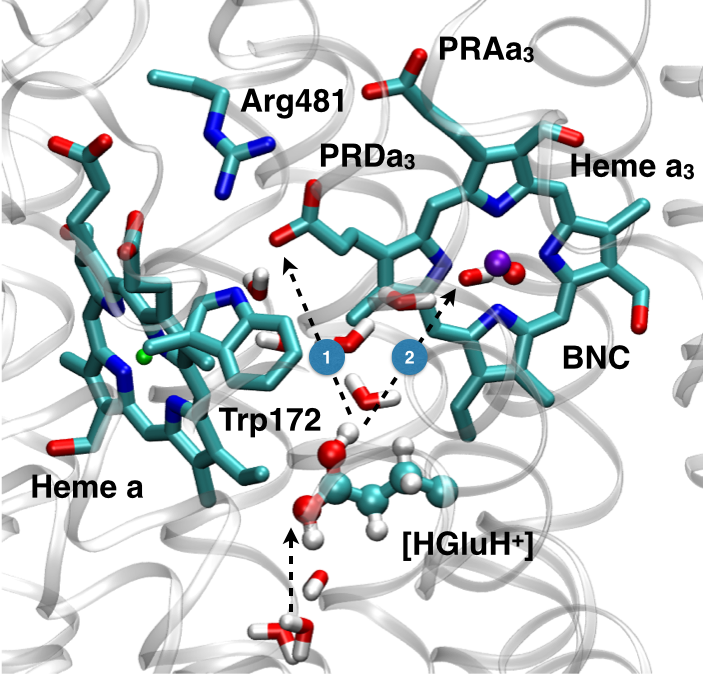
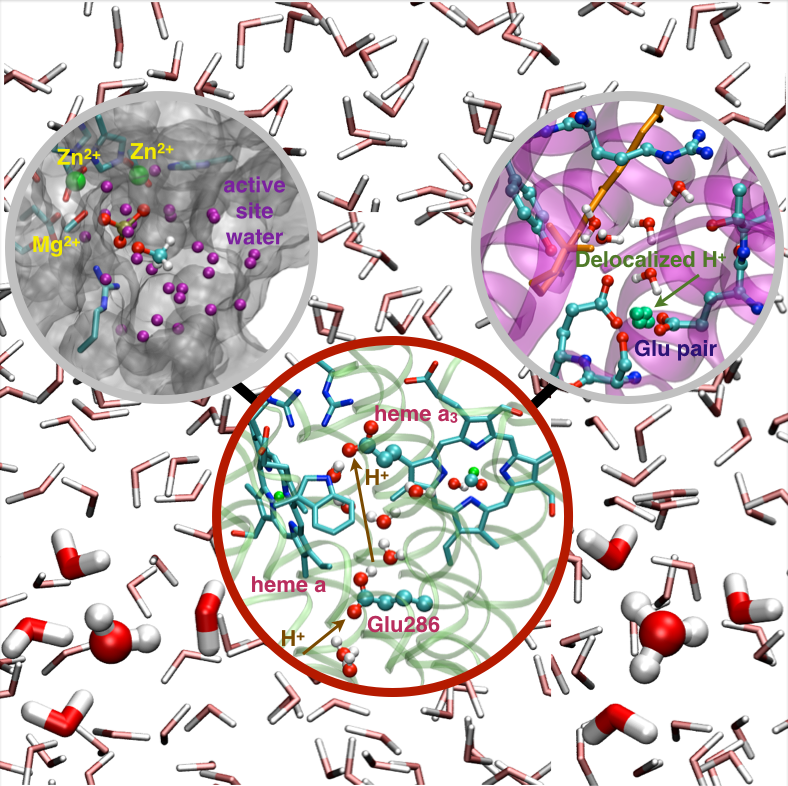
Microscopic basis for kinetic gating in CcO:
Understanding the mechanism of vectorial proton pumping in biomolecules requires establishing the microscopic basis for the regulation of both thermodynamic and kinetic features of the relevant proton transfer steps. Using DFTB3/MM simulations, we show that protonation of the proton loading site (PLS) in CcO requires a concerted process in which a key glutamic acid (Glu286H) delivers the proton to the PLS while being reprotonated by an excess proton coming from the D-channel. The concerted nature of the mechanism is a crucial feature that enables the loading of the PLS before the cavity containing Glu286 is better hydrated to lower its pK to experimentally measured range. In addition, we find that rotational flexibility of the PLS allows its protonation before that of the binuclear center. This work provides microscopic support to the kinetic constraints revealed by kinetic network analysis as essential elements that ensure an efficient vectorial proton transport in cytochrome c oxidase. Chem. Sci., 6, 826-841 (2015)
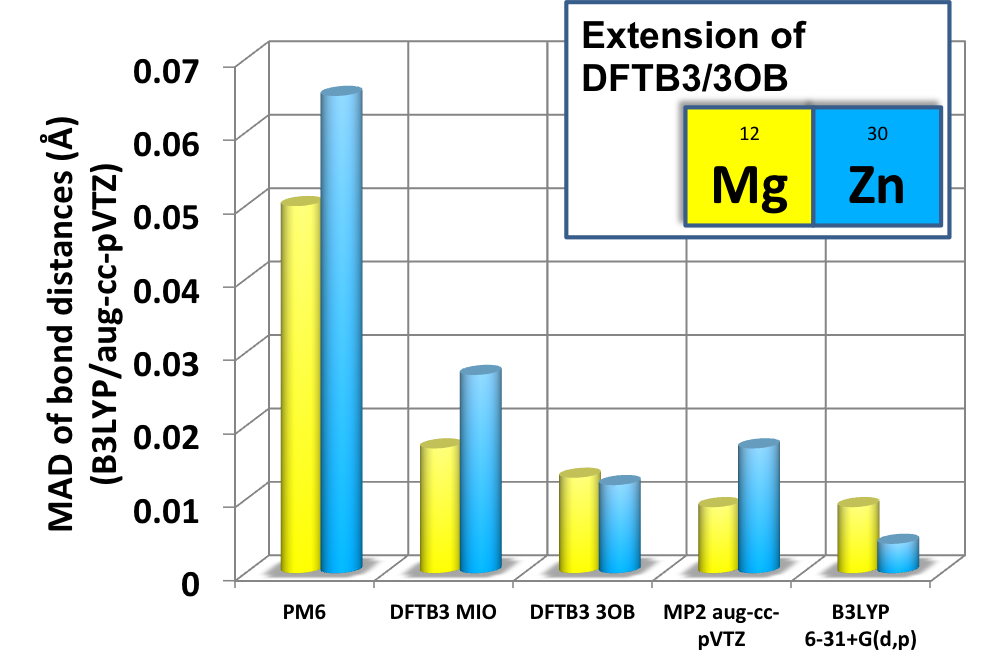
DFTB3/3OB for Magnesium and Zinc:
We report the parameterization of the approximate density functional theory, DFTB3, for Magnesium and Zinc for chemical and biological applications. The parameters are benchmarked using both gas phase and condensed phase systems. The results indicate that DFTB3/3OB is particularly successful at predicting structures, including rather complex dimension-nuclear metalloenzyme active sites, while being semi-quantitative (with a typical MAD of ~3-5 kcal/mol) for energetics. Single point calculations with high-level QM methods generally lead to very satisfying (a typical MAD of ~1 kcal/mol) energetic properties. J. Phys. Chem. B (Jorgensen Issue) , 119,1062-82 (2015)
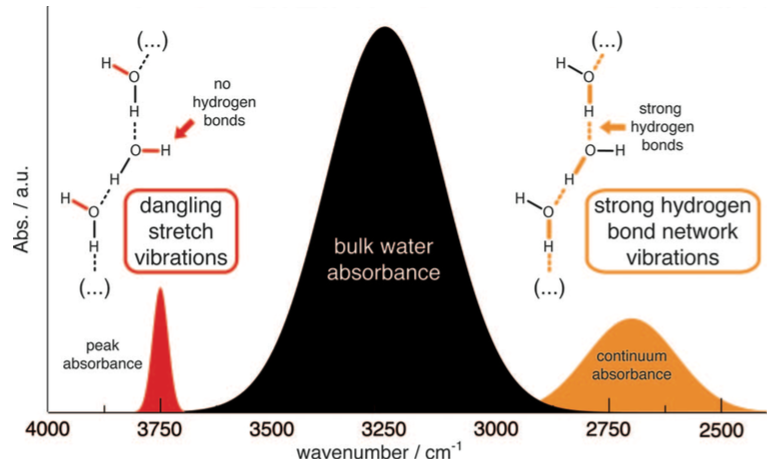
Resolving the structure of transient water wires in protein:
Transient water wires/clusters play an important role in the function of many proteins. The composition and structure of such clusters/wires are difficult to resolve using crystallography or NMR. By combining time-resolved infrared difference spectroscopy and QM/MM simulations, we and the Gerwert group are able to resolve the identity and spectroscopic signature of a water wire buried in the membrane protein bacteriorhodopson. This methodology is expected to be applicable to study functionally relevant water dynamics in proteins in general. J. Chem. Phys., 141, 22D524 (2014)
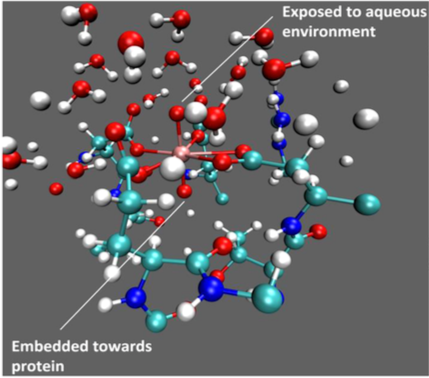
Understanding uranyl binding selectivity in protein:
Understanding the physical basis for metal binding selectivity by proteins is of both fundamental and practical implications. Molecular dynamics and free energy calculations are used in this study to probe the capture of uranyl by a protein recently designed by the He and Lai groups. The roles of active site residues and water in determining the binding affinity and selectivity are made clear by the simulations. Moreover, a mutant was designed with further enhanced binding affinity & selectivity, which was qualitatively supported by new experiments. This joint study highlights that even for a protein with femto-molar binding affinity, computational analysis can help improve the binding properties. J. Am. Chem. Soc., 136, 17484-17494 (2014)
DFTB3 description of water and hydration effects in different environments:
In this article we discuss the description of water and hydration effects that employs an approximate density functional theory, DFTB3, in either a full QM or QM/MM framework. The goal is to explore, with the current formulation of DFTB3, the performance of this method for treating water in different chemical environments, the magnitude and nature of changes required to improve its performance, and factors that dictate its applicability to reactions in the condensed phase in a QM/MM framework. By comparing results from DFTB3 models that differ in the description of water, we confirm that proton transfer energetics are adequately described by the standard DFTB3/3OB model for meaningful mechanistic analyses. For QM/MM applications, we emphasize that a robust parameterization of QM-MM interactions requires an explicit consideration of condensed phase properties. J. Phys. Chem. B Feature Article , 118, 11007-11027 (2014)
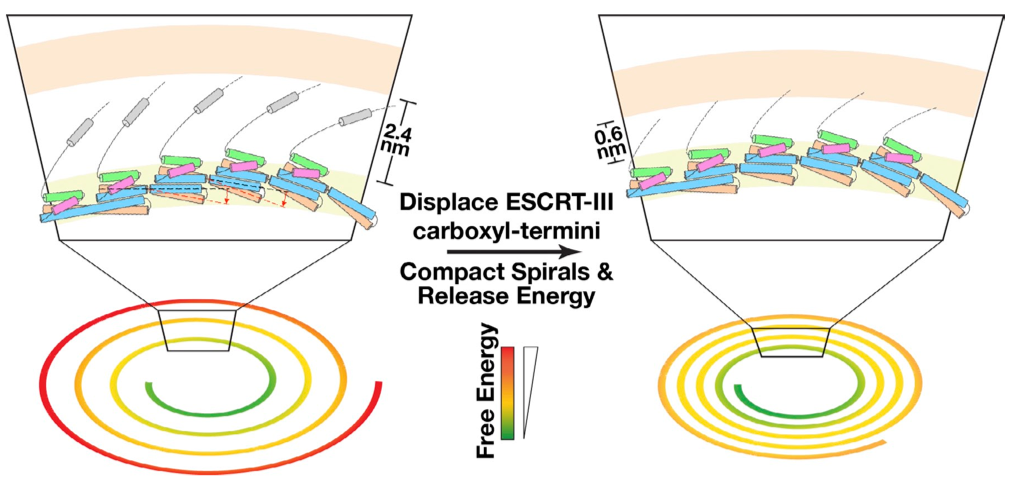
Mechanisms governing ESCRT-III spiral filament assembly:
ESCRT-III (endosomal sorting complex required for transport III) filaments mediate mem- brane scission during the ostensibly disparate processes of multivesicular endosome biogenesis, cytokinesis, and retroviral budding. However, mechanisms by which ESCRT-III subunits assemble into a polymer remain unknown. Using a combination of cryo-Electron Microscopy, molecular dynamics simulations and site-directed mutagenesis, we identified the structural flexibility and protein-protein interactions that underlie the assembly of ESCRTIII filaments, leading to a new hypothesis regarding how these filaments drive membrane fission. J. Cell Biol., 206, 763-777 (2014)
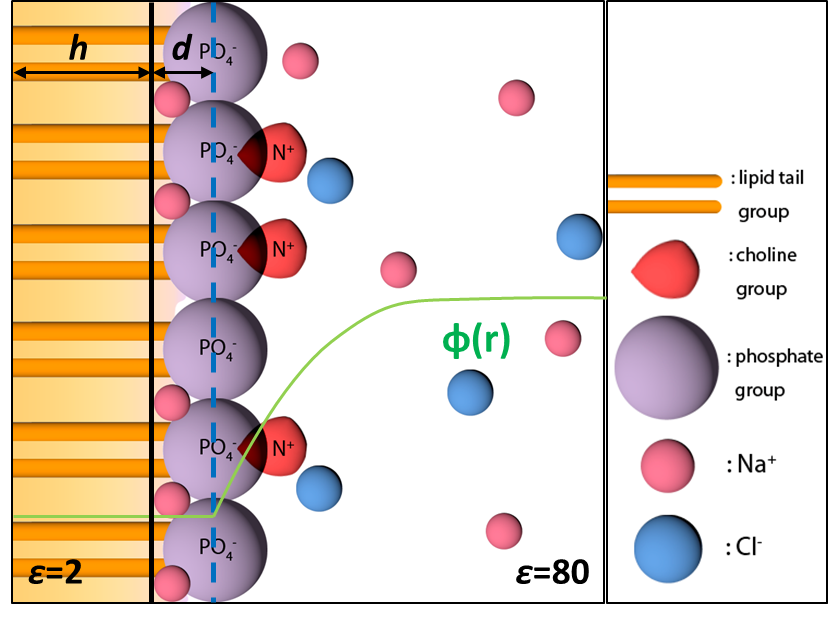
Free Energy Calculations for Peripheral Binding of Proteins to Membrane:
The binding of peptides and proteins to the surface of complex lipid membranes is important in many biological processes such as cell signaling and membrane remodeling. In this work, we explore a method based on an implicit membrane/solvent model (generalized Born with a simple switching in combination with the Gouy-Chapman-Stern model for a charged interface), which we expect to lead to useful results when the binding does not implicate significant membrane deformation and local demixing of lipids. We show that the binding free energy can be efficiently computed following a thermodynamic cycle similar to protein−ligand binding calculations, especially when a Bennett acceptance ratio based protocol is used to consider both the membrane bound and solution conformational ensembles. The calculations also highlight that for a membrane with a significant fraction of anionic lipids, it is essential to include the effect of ion adsorption using the Stern model, which significantly modifies the effective surface charge. J. Chem. Theo. Comp., 10, 2845-2859 (2014)
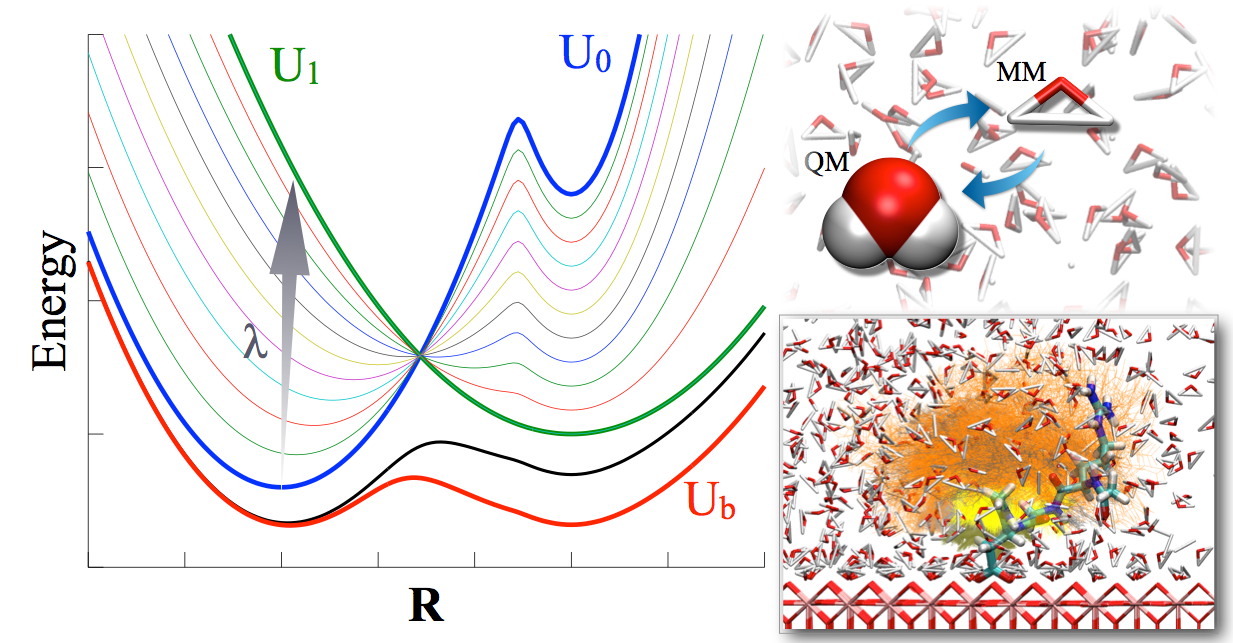
An integrated Hamiltonian approach for sampling:
Motivated by the recent work of Gao and co-workers on integrated tempering sampling (ITS), we have developed a novel sampling approach referred to as integrated Hamiltonian sampling (IHS). The method carries out sampling using an effective Hamiltonian constructed by integrating the Boltzmann distributions of a series of Hamiltonians. By judiciously selecting the weights of the different Hamiltonians, one achieves rapid transitions among the energy landscapes that underlie different Hamiltonians and therefore an efficient sampling of important regions of the conformational space. Along this line, IHS shares similar motivations as the enveloping distribution sampling (EDS) approach of van Gunsteren and co-workers, although the ways that distributions of different Hamiltonians are integrated are rather different in IHS and EDS. Specifically, we report efficient ways for determining the weights using a combination of histogram flattening and weighted histogram analysis approaches, which make it straightforward to include many end-state and intermediate Hamiltonians in IHS so as to enhance its flexibility. J. Phys. Chem. B (Skinner issue), 118, 8210-8220 (2014)
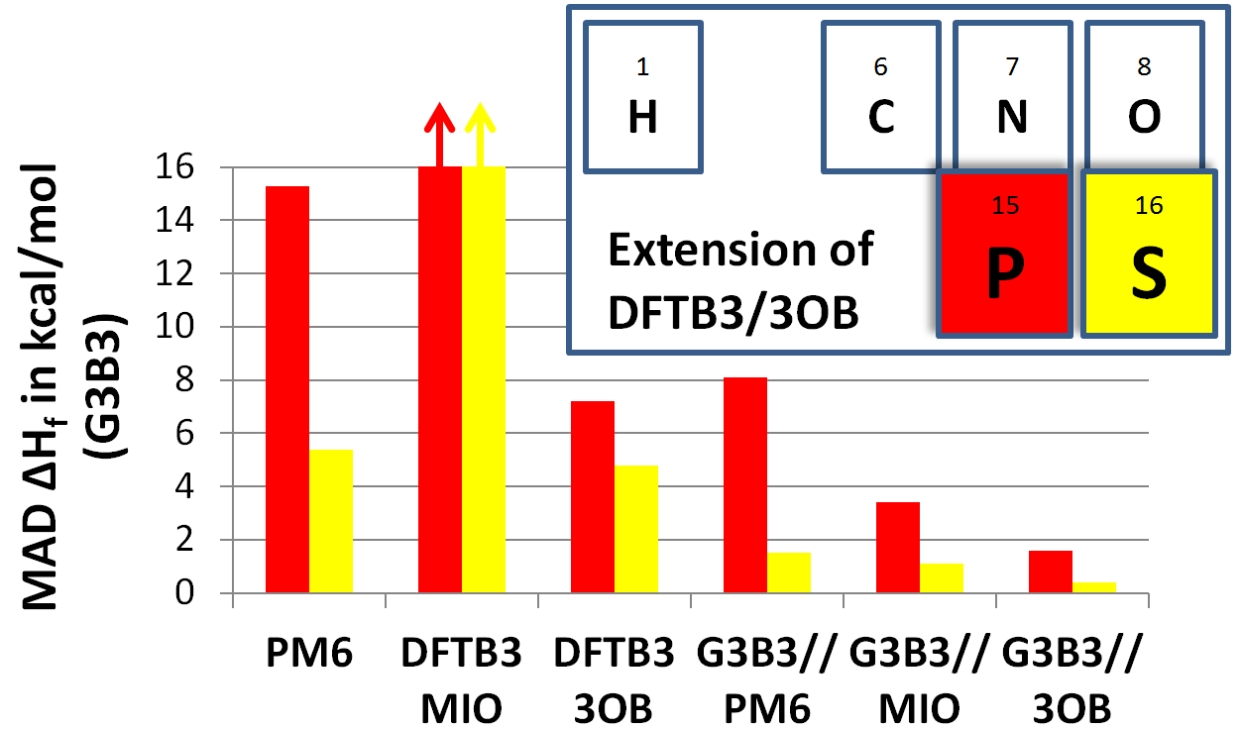
DFTB3/3OB for Sulfur and Phosphorus:
We report the parameterization of DFTB3, for sulfur and phosphorus in a framework of the 3OB set. The 3d orbitals are included in the parameterization and the electronic parameters are chosen to minimize errors in the atomization energies. In general, DFTB3/3OB is a major improvement over the previous parameterization (DFTB3/MIO), and for the majority cases tested, it also outperforms PM6 and PDDG, especially for structural properties, vibrational frequencies, hydrogen bonding interactions and proton affinities. Despite specific limitations, DFTB3/3OB is expected to be a competitive QM method in QM/MM calculations for studying phosphorus/sulfur chemistry in condensed phase systems, especially as a low-level method that drives the sampling in a dual-level QM/MM framework. J. Chem. Theo. Comp., 10, 1518-1537 (2014)
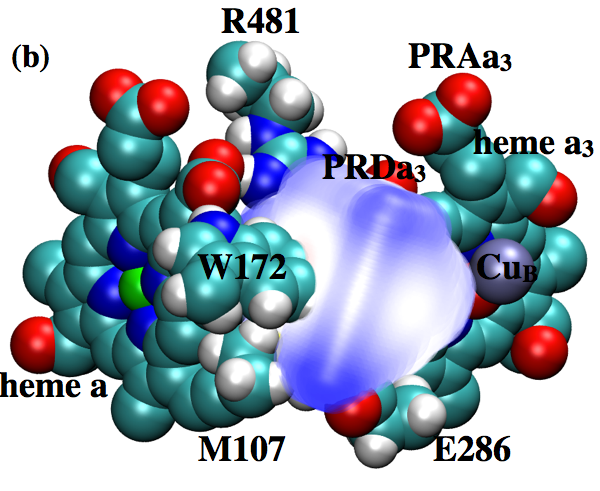
Changing hydration level of a protein cavity is functionally important:
Cytochrome c oxidase is an important proton pump that utilizes the chemical energy released by oxygen reduction to generate the transmembrane proton concentration gradient. A conserved Glutamate residue has been proposed to play a key role in proton pumping , although factors that control the timing and destination of proton transfers by this residue remain poorly understood. By integrating results from multiple computational methodologies, we propose a novel mechanism in which changes in local hydration and electrostatic interactions regulate the proton affinity of this key residue and, as a result, proton transfer activities. The results highlight the functional significance of local protein motions and hydration state of internal cavities. Proc. Natl. Acd. Sci. , 110, 18886-18891 (2013)
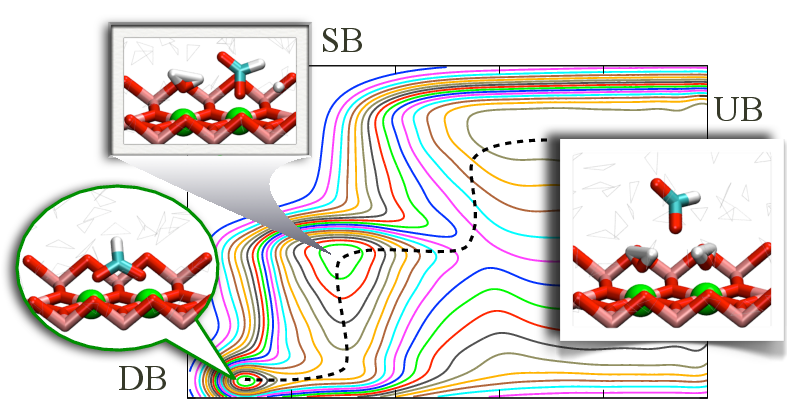
Desolvation as a critical step for adsorption at ionic surface:
Understanding factors that control the affinity and specificity of biomolecule-nanoparticle interactions is essential to the study of many phenomena such as biomineralization and cell uptake of nano-materials. To establish an efficient and effective computational protocol for such problem, we have studied the binding/unbinding of a formate anion to the rutile TiO_2 (110) surface. Our results (metadynamics and alchemical free energy perturbation) clearly demonstrate that a reliable description of the binding process requires an explicit consideration of changes in the solvation state of the binding site. The insights and methods established here are generally applicable to the analysis of solid/liquid interface. J. Chem. Theo. Comp., 9, 5059-5069 (2013)
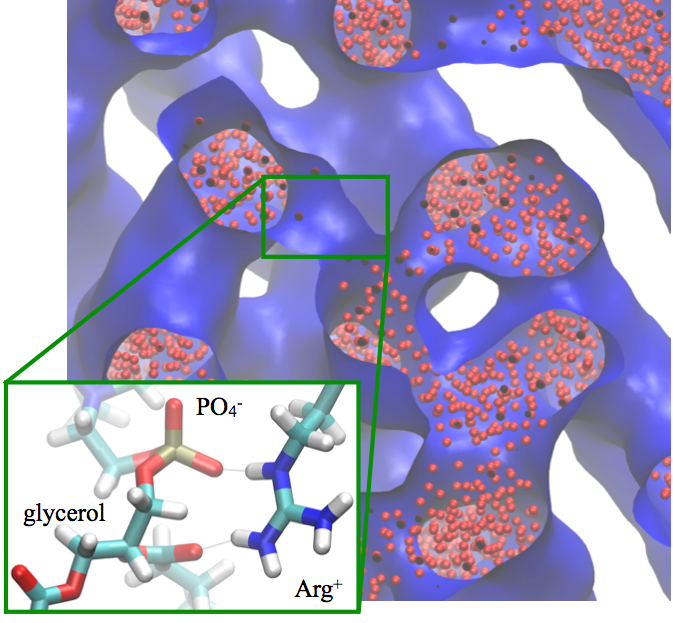
The difference between Arg and Lys in interaction with lipids:
An important puzzle in membrane biophysics is the difference in the behaviors of Lysine (Lys) and Arginine (Arg) based peptides at the membrane. For example, the translocation of poly-Arg is orders of magnitude faster than that of poly-Lys; when mixed with lipids, poly-Arg and poly-Lys form very different phases. Using atomistic and CG models, we show that electrostatic and glycerol-peptide interactions play a crucial role in determining the phase behavior of peptide-lipid mixtures, with the difference between Arg and Lys arising from the stronger interactions of the former with lipid glycerols. In other words, the multivalent nature of the guanidinium group allows Arg to simultaneously interact with both phosphate and glycerol groups, while Lys engages solely with phosphate; this feature of amino acid/lipid interactions has not been emphasized in previous studies. J. Phys. Chem. B , 117, 12145-12156 (2013)
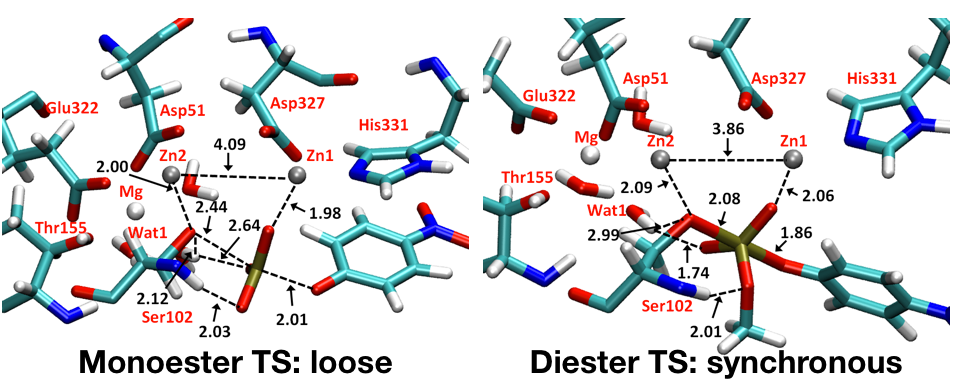
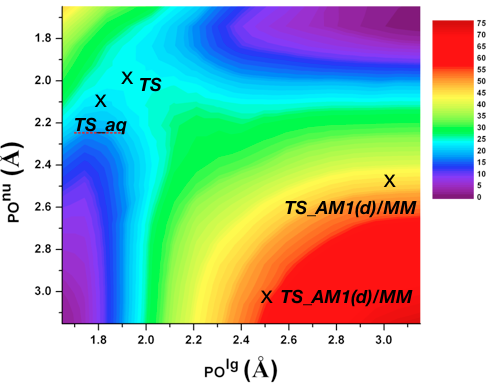
A single enzyme active site stabilizes multiple types of transition states:
Conventional picture for enzyme catalysis implies that enzyme active sites have evolved to stabilize a specific type of transition state. QM/MM analysis of phosphoryl transfer reactions in the alkaline phosphatase (AP) enzymes explicitly supports the proposal that these enzymes are able to recognize and stabilize different types of transition states in a single active site. Analysis of the structural features of computed transition states indicates that the plastic nature of the bimetallic site plays a minor role in accommodating multiple types of transition states and that the high degree of solvent accessibility of the AP active site also contributes to its ability to stabilize diverse transition-state structures without the need of causing large structural distortions of the bimetallic motif. J. Am. Chem. Soc. , 135, 10457-10469 (2013)
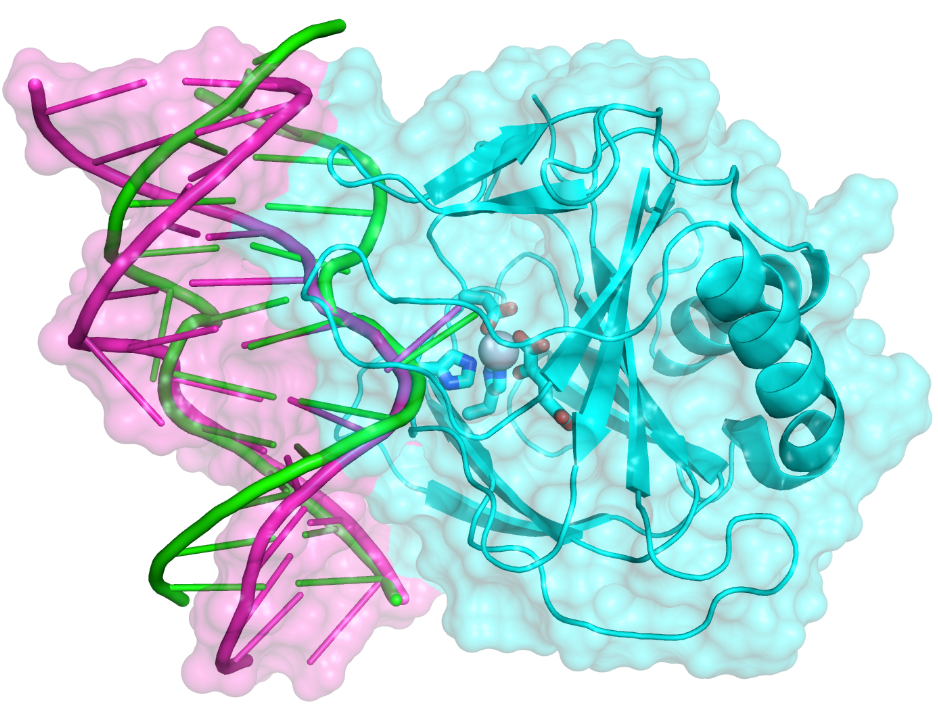
A simple MM model for Fe(II) and application to AlkB enzymes:
A simple molecular mechanical model for Fe(II) has been developed and tested with 150 ns MD simulations for enzymes in the AlkB family. Although the structural features for the ABH2-dsDNA complex are overall in good agreement with the crystal structure, the dsDNA and AlkB-dsDNA interface undergo substantial changes during the MD simulations from the crystal structure. The results highlight that crystal packing may have a significant impact on the structure of protein-DNA complexes; the simulations also provide additional insights regarding why AlkB and ABH2 prefer single-strand and double-strand DNA, respectively, as substrate. J. Comp. Chem. , 34, 1620-1635 (2013)
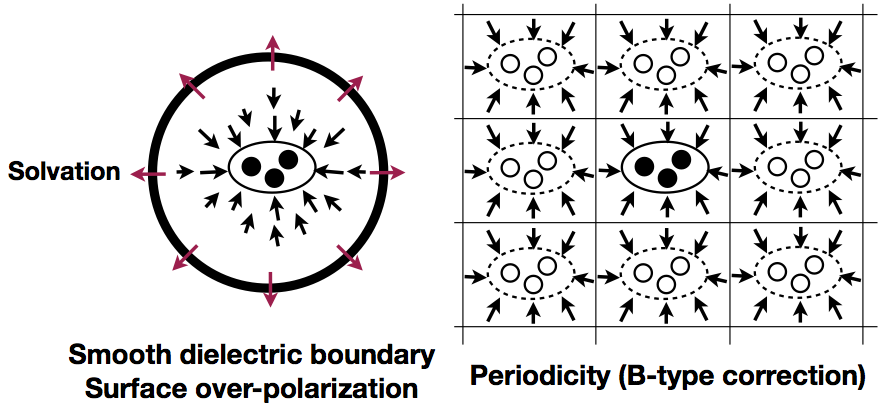
Analysis of charging free energy simulations with PBC and GSBP:
Free energy simulations using a finite sphere boundary condition rather than a periodic boundary condition (PBC) are attractive in the study of very large biomolecular systems. To understand the quantitative impact of various approximations in such simulations, we compare charging free energies in both solution and protein systems calculated with the Generalized Solvent Boundary Potential (GSBP) and PBC simulations. The results suggest that GSBP and PBC charging free energies generally agree, once the relevant correction terms are taken into consideration. J. Phys. Chem. B, 117, 2005-2018 (2013)
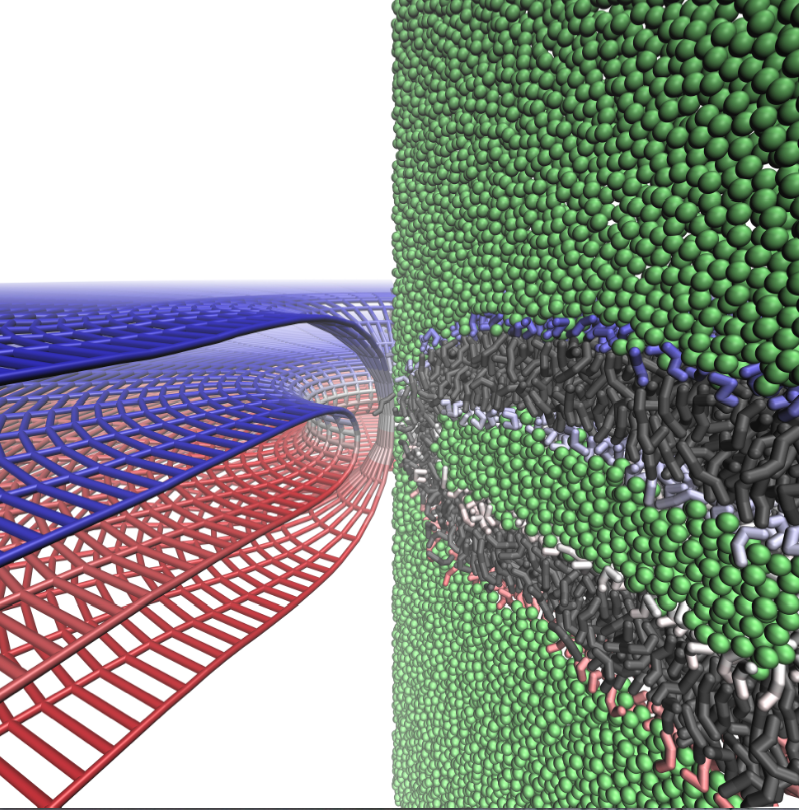
Continuum mechanics vs. coarse-grained models for the membrane fusion pore:
To establish the validity of continuum mechanics models quantitatively for the analysis of membrane remodeling processes, we compare the shape and energies of the membrane fusion pore predicted by coarse-grained (MARTINI) and continuum mechanics models. The results at these distinct levels of resolution give surprisingly consistent descriptions for the shape of the fusion pore, and the deviation between the continuum and coarse-grained models becomes notable only when the radius of curvature approaches the thickness of a monolayer. These observations provide solid support for the use of coarse-grained and continuum models in the analysis of membrane remodeling. The combined coarse-grained and continuum analysis confirms the recent prediction of continuum models that the fusion pore is a meta-stable structure and that its optimal shape is neither toroidal nor catenoidal. Moreover, our results help reveal a new `bowing’ feature in which the bilayers close to the pore axis separate from one another more than at greater distances from the pore axis; bowing helps reduce the curvature and therefore stabilizes the fusion pore structure. J. Yoo et al. Biophys. J., 104, 841-852 (2013)

Analysis of stress profiles around membrane proteins & protein-protein interactions:
It’s been increasingly recognized that mechanical properties of biomembranes can significantly modulate the functional characteristics of membrane proteins. To better understand the underlying physical basis and construct predictive models for such modulation, we use both atomistic and coarse-grained simulations to compute stress field distribution around a membrane protein using gA dimer as a model system. Moreover, we carry out such calculations with lipid bilayers of different thickness to probe how hydrophobic mismatch influences the stress field. Finally, we use coarse-grained models to study the association of multiple gA dimers and analyze the results using stress profile calculations. Collectively, the results quantitatively demonstrate the importance of the protein/membrane interface and help highlight both the value and limitation of a mechanical perspective. J. Yoo et al. Biophys. J., 104, 128-138 (2013)
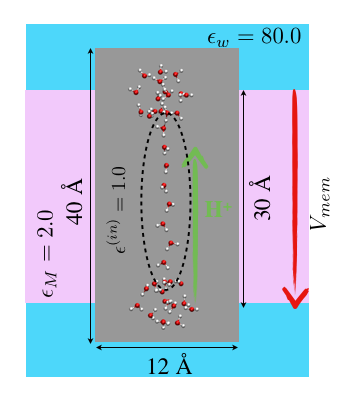
Implementation of SMBP for ab initio QM/MM in CHARMM:
Although the generalized solvent boundary potential (GSBP) has proven to be a useful approach for carrying out efficient MD simulations with semi-empirical QM/MM (e.g., SCC-DFTB/MM) potentials, its implementation is more involved with ab initio QM methods. As an alternative for geometry minimization applications (e.g., reaction path search), we have implemented the SMBP into the program package CHARMM with the ab-initio quantum mechanical program packages Q-Chem and Gaussian. We have further demonstrated the robustness of the surface charge approach to represent the electrostatic interactions with the QM part due to the boundary potential, and shown that the SMBP is also robust with respect to the choice of the atomic QM (ESP) charges. The SMBP is currently the only general boundary potential approach that can be coupled with ab-initio QM/MM methods. One unique aspect of our implementation is that it is able to analyze the influence of membrane potential on chemical reactions in transmembrane proteins. J. Zienau et al. J. Phys. Chem. B, 116, 12522-12534 (2012)
A new QM/MM electrostatic model for SCC-DFTB:
QM/MM methods have become indispensable in the analysis of reaction mechanisms of complex systems. Once specific QM and MM models are chosen for a given application, the remaining factor that determines the accuracy of the calculation is the coupling between the QM and MM models. Motivated by models used to approximate two electron integrals in semi-empirical QM methods, we propose to revise the electrostatic part of the SCC-DFTB/MM interaction to adopt a Klopman-Ohno (KO) functional form, which mimics the interaction between two spherical charges; this takes charge penetration effects into consideration and therefore significantly improves the description of charge-charge interactions at short range. To be consistent with the third-order formulation of SCC-DFTB, the Hubbard parameter in the KO functional is dependent on the QM charge. In this way, the effective size of the QM charge distribution naturally adjusts as the QM region undergoes chemical transformations, making the KO based QM/MM scheme particularly attractive for describing chemical reactions in the condensed phase. G. Hou et al. J. Chem. Theo. Comp., 8, 4293-4304 (2012)
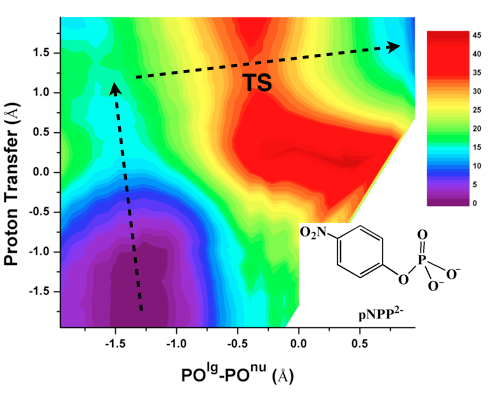
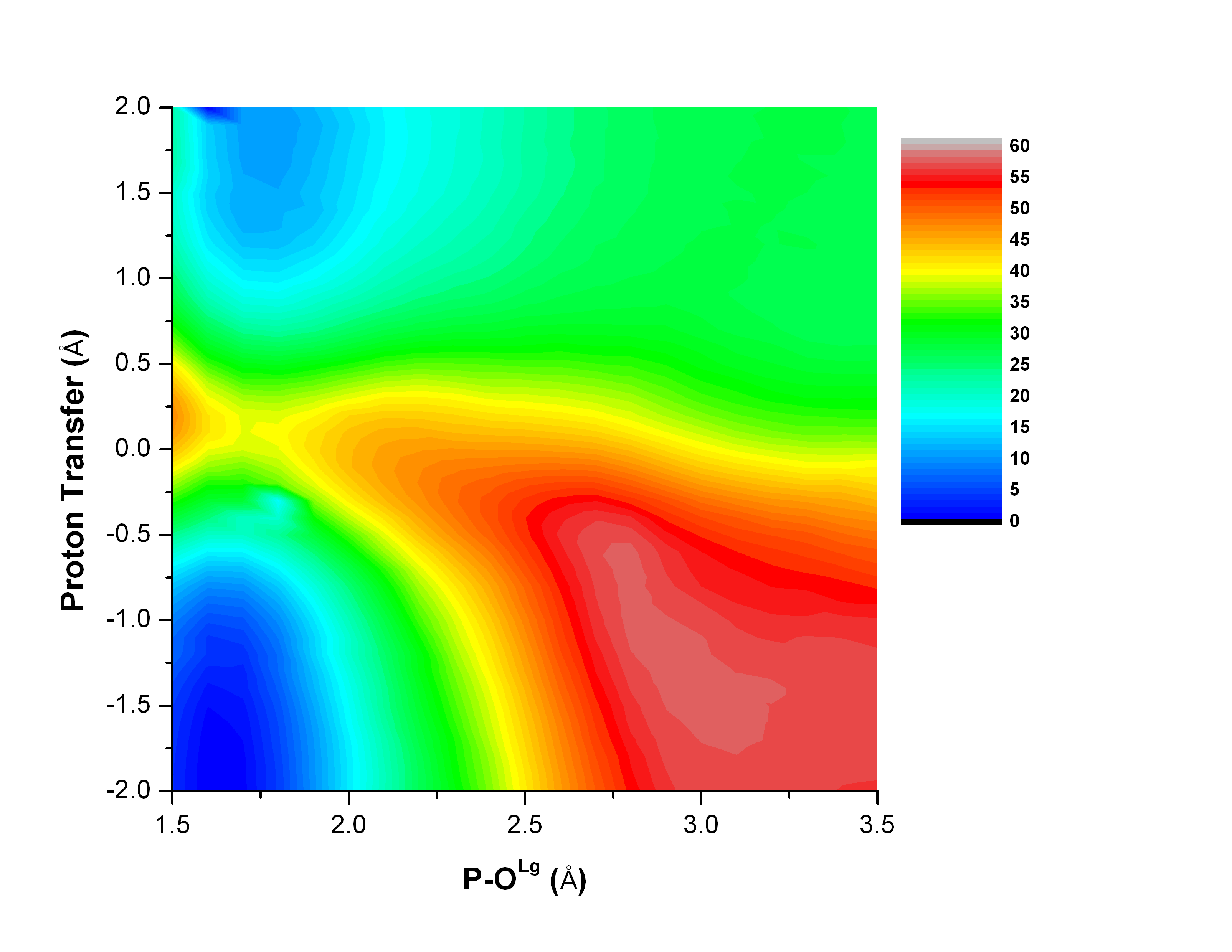
Catalytic promiscuity and phosphoryl transfer transition state:
Several members of the Alkaline Phosphatase (AP) superfamily exhibit a high level of catalytic proficiency and promiscuity in structurally similar active sites. A thorough characterization of the nature of transition state for different substrates in these enzymes is crucial for understanding the molecular mechanisms that govern those remarkable catalytic properties. Using calibrated SCC-DFTBPR/MM simulations, we demonstrate that hydrolysis of phosphate diesters catalyzed by AP and NPP feature similar synchronous transition states that are slightly tighter in nature compared to that in solution. Therefore, this study provides the first direct computational support to the hypothesis that enzymes in the AP superfamily catalyze cognate and promiscuous substrates via similar transition states to those in solution. G. Hou et al. JACS, 134, 229 (2012)
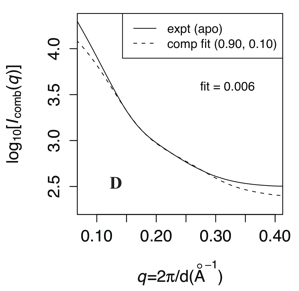
Conformational ensemble in solution:
While coarse-grained (CG) simulations provide an efficient approach to identify small- and large-scale motions important to protein conformational transitions, coupling with appropriate experimental validation is essential. Here, by comparing small-angle X-ray scattering (SAXS) predictions from CG simulation ensembles of adenylate kinase (AK) with a range of energetic parameters, we demonstrate that AK is flexible in solution in the absence of ligand and that a small population of the closed form exists without ligand. By analyzing variation of scattering patterns within CG simulation ensembles, we reveal that rigid-body motion of the LID domain corresponds to a dominant scattering feature. Thus, we have developed a novel approach for 3D structural interpretation of SAXS data. M. Daily et al. Chem. Phys. 396, 84 (2012)
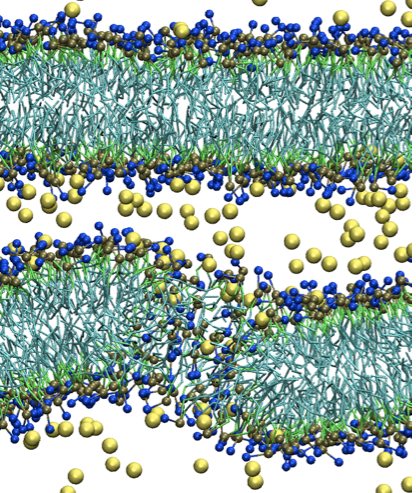
A new CG model for membrane-peptide simulations:
We present a new coarse-grained (CG) model for simulations of lipids and peptides. The model follows the same topology and parameterization strategy as the MARTINI force field, but is based on our recently developed big multipole water (BMW) model for water. The new BMW-MARTINI force field reproduces many fundamental membrane properties and also yields improved energetics (when compared to the original MARTINI force- field) for the interactions between charged amino acids with lipid membranes, especially at the membrane/water interface. The BMW-MARTINI model is particularly suitable for describing interactions between highly charged peptides with lipid mem- branes, which is crucial to the study of antimicrobial peptides, cell penetrating peptides, and other proteins/peptides involved in the remodeling of biomembranes. Z. Wu et al. JCTC, 11,3793 (2011)
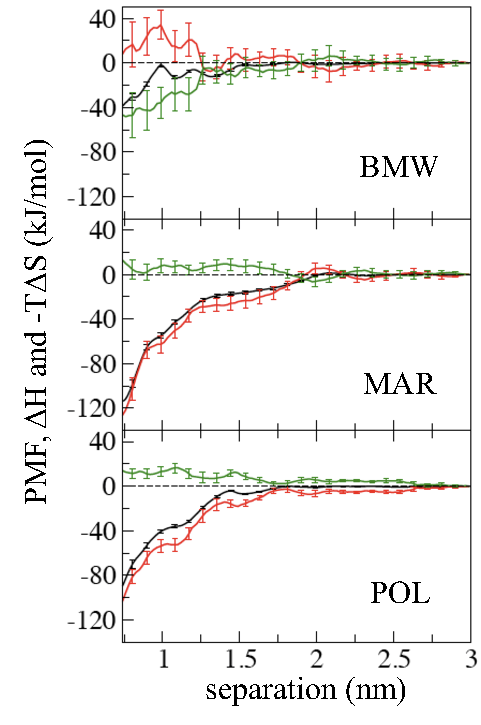
Importance of water model in CG simulations:
The hydrophobic effect plays a central role in many biological processes including protein folding and aggregation. The hydrophobic interaction between solutes, such as helical peptides, is believed of entropic origin, and driven by the increase in the entropy of water due to association. In this work we examine the association between peptides in water using several coarse-grained (CG) models, such as MARTINI (MAR), Polarizable MARTINI (POL) and Big Multipole Water (BMW) models. All models predict that a pair of helical peptides (Ala20 and Leu20) has favorable association free energy. The BMW model is the only model, however, in which this association is entropy driven, as has been previously established with atomistic simulations and experiments. The MAR and POL models, where the CG water particles do not have a quadrupole moment, predict an enthalpy driven association, with a negligible entropy change upon association. These results emphasize the importance of electrostatic interactions in water for the qualitative features of thermodynamics of biophysical systems. Z. Wu et al. JPC-Lett. 2, 1794 (2011)
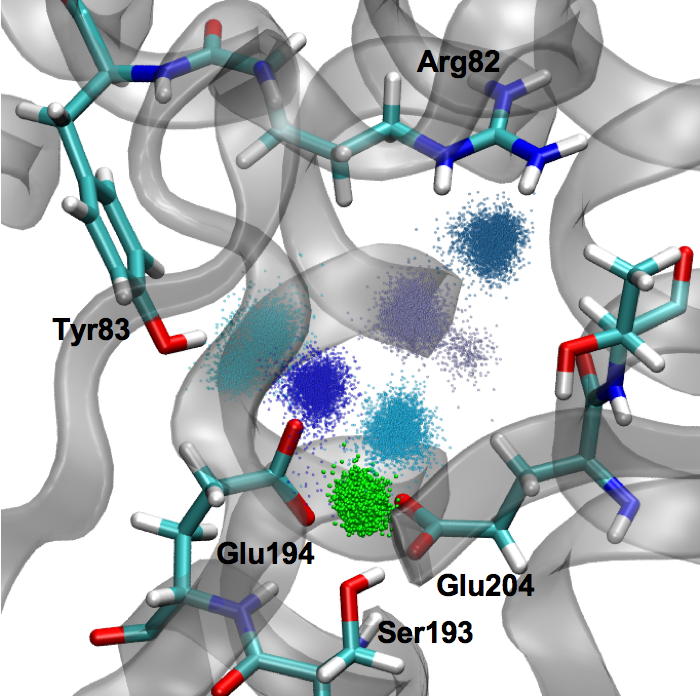
pKa, water distribution of the proton loading site in bR:
Identifying the group that acts as the proton storage/loading site is a challenging but important problem for understanding the mechanism of proton pumping in biomolecular proton pumps. Recent experimental studies of bacteriorhodopsin (bR) propelled the idea that the proton storage/release group (PRG) in bR is not an amino acid but a water cluster embedded in the protein. We argue that this idea is at odds with our knowledge of protein electrostatics, and our recent QM/MM simulations suggested an alternative “intermolecular proton bond” model in which the stored proton is shared between two conserved Glu residues. Here we show that this model leads to microscopic pKa values consistent with available experimental data and the functional requirement of a PRG. The results and analyses help highlight the importance of considering protein electrostatics and provide arguments for why the “intermolecular proton bond” model is likely applicable to PRG in biomolecular proton pumps in general. Goyal et al., J. Am. Chem. Soc. 133, 14981-14997 (2011)
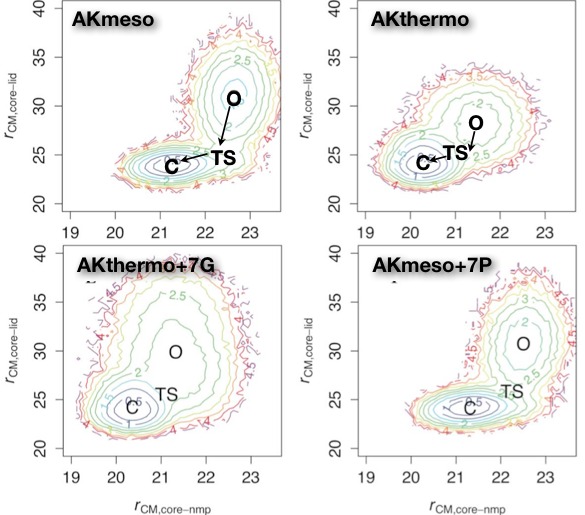
Interconversion of Functional Motions between homologous proteins:
Dynamic properties are functionally important in many proteins, including the enzyme adenylate kinase (AK), which undergoes chemically rate-limiting domain motions coupled to substrate binding. Since mesophiles and thermophiles often differ in functionally important motions, we compare coarse-grained simulations of AKmeso and AKthermo as well as several proline and glycine mutational variants designed to interconvert the dynamics. As might be expected, both domain motions and local unfolding motions are reduced in AKthermo relative to AKmeso. In AKthermo, both of these types of motions can be partially shifted toward more flexible AKmeso by heating or by mutating hinge prolines. However, only mutation to highly flexible glycine produces motions like those of AKmeso. Thus, the rate-limiting global transition likely depends on a combination of hinge flexibility and stability within the LID and NMP domains. DDaily et al., PLoS Comp. Biol. 7, e1002103 (2011)
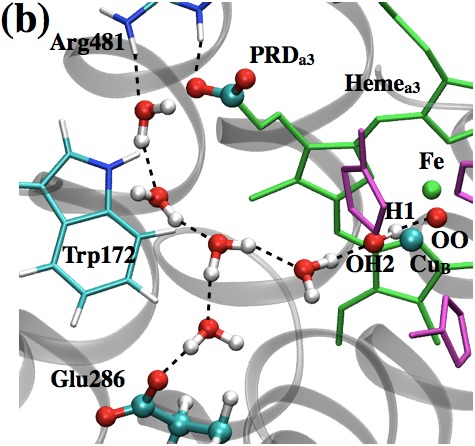
Active site properties of Cytochrome c oxidase:
One of the key unsolved issues regarding proton pumping in Cytochrome c Oxidase (CcO) concerns the identity of the gating element(s) that prevents the backflow of protons. In this study, we analyze two popular proposals for the gating elements: isomerization of the key branching residue, Glu 286, and (re)orientation of water molecules in the hydrophobic cavity. Using a multi-faceted set of computational analyses that involve CcO embedded in either an implicit or explicit treatment of lipid membrane, we show that neither Glu 286 nor active site water likely constitutes the gating element. Yang et al., Biophys. J. 101, 61-69 (2011)
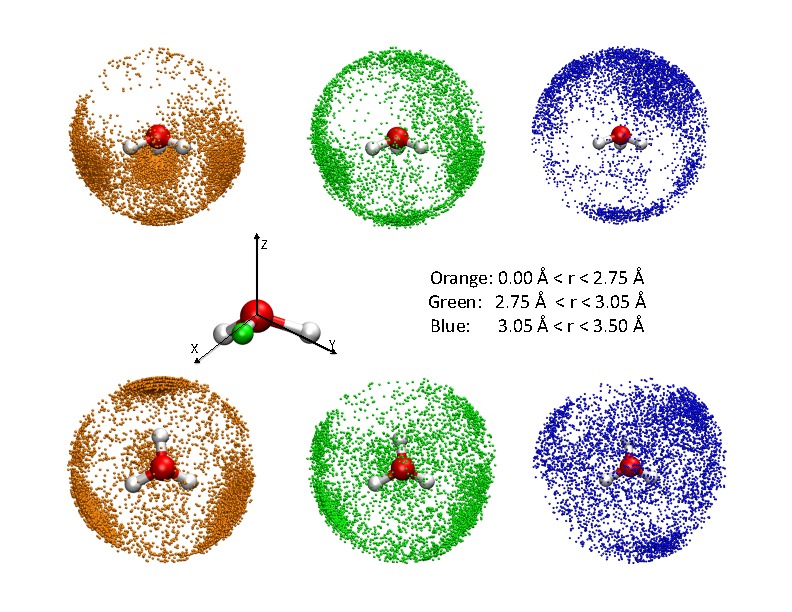
SCC-DFTB applied to water clusters and bulk water
Since water is arguably the most important solvent in chemistry and (certainly) in biology, it is useful to understand the applicability and limitations of approximate quantum mechanical methods when applied to water. In this work, we explore the performance of SCC-DFTB for protonated gas-phase water clusters and bulk water (the latter both with and without an excess proton) with a modified O-H repulsive potential reported in our earlier work and with on-site third-order expansion of the DFT energy. Our results show that, with the proper set of published parameters, SCC-DFTB does correctly favor the Eigen form of the hydrated proton compared to the Zundel form, both in gas-phase clusters and in the bulk; the amphiphilic character of the hydrated proton discussed in the literature has also been observed. Goyal et al., J. Phys. Chem. B 115, 6790 (2011)
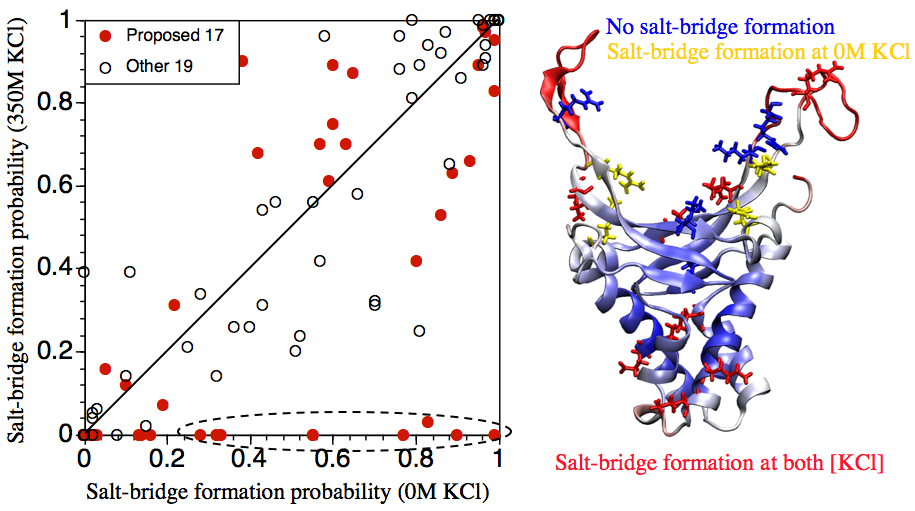
Salt-bridge dynamics in DNA wrapping proteins:
Disruption of salt-bridges have been proposed, based on thermodynamic and structural analyses, to be involved in the binding of protein-wrapping proteins, such as IHF, to DNA. Since the apo structure of IHF is not available (due presumably to its flexibility), we use MD simulations starting from the IHF conformation in IHF-DNA complex to explore whether salt-bridge dynamics are likely functionally relevant. The results suggest that, from a structural point of view, the proposal is plausible. However, the complex behaviors of charged residues observed in the MD simulations also suggest that the unusual thermodynamic characteristics for IHF-DNA binding likely arise from the interplay between complex dynamics of charged residues both in and beyond the DNA binding site. Ma et al., Biochem. 50, 266 (2011)
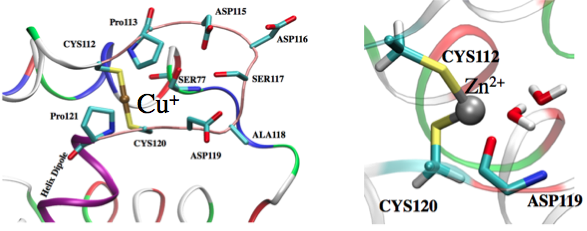
Factors that regular metal binding specificity:
Factors that regulate the binding affinity and selectivity of proteins to transition metal ions remain poorly understood. Here we develop a new QM/MM and Poisson-Boltzmann based approach to systematically dissect contributions from different factors in the Copper efflux Regulator (CueR). Our analysis highlights both intrinsic properties of a metal ion and protein contributions. Specifically, the softness and desolvation penalty of a metal ion make large contributions to the binding affinity; for example, we find that the large desolvation penalty for Zn2+ rather than any stereoelectronic factor (e.g., linear vs. tetrahedron coordination) is the key reason that Zn2+ binds much more weakly than Hg2 to CueR. Moreover, our results explicitly demonstrate that the electrostatic environment of CueR is well-tuned to favor the binding of coinage metal ions over divalent ions. Rao et al., J. Am. Chem. Soc. 132, 18092 (2010)
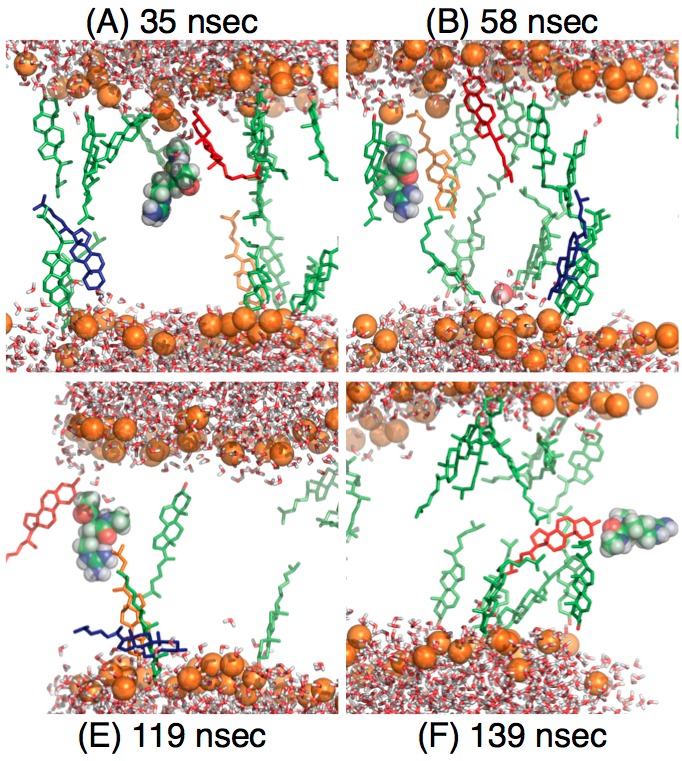
Effect of cholesterol on the pKa of Arg in a lipid membrane:
Extensive free energy simulations of microscopic pKa of an Arg analogue in the membrane center indicate that cholesterol molecules decrease the pKa of Arg not by increasing the energy penalty for membrane deformation (water defect formation) in the presence of charged state of Arg. Instead, cholesterol molecules stabilize the neutral form of Arg. Therefore, while mechanical considerations are useful for stimulating hypothesis, their applicability to dissecting phenomena at the molecular length scale is rather limited. Yoo et al., Biophys. J. 99, 1529-1538 (2010)
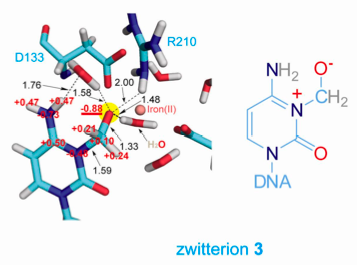
QM/MM calculations indicate that a critical intermediate in AlkB is zwitterionic:
E. coli AlkB is a prototype of iron-containing dioxygenases that catalyze various oxidative demethylation of nucleic acids and histones. Employing a chemical cross-linking strategy, complexes of AlkB-dsDNA containing several substrate analogues were stabilized and crystallized. Exposing these crystals, grown under anaerobic conditions, to dioxygen initiates oxidation in crystallo, which led to the capture of several key intermediates. The direct observation and QM/MM characterization of these otherwise unstable chemical intermediates provides the validation of the oxidative demethylation pathway proposed for this family of enzymes, and also offers insights into the general demethylation mechanism used by these demethylases. See, Yi et al., Nature , 468, 330-333 (2010)
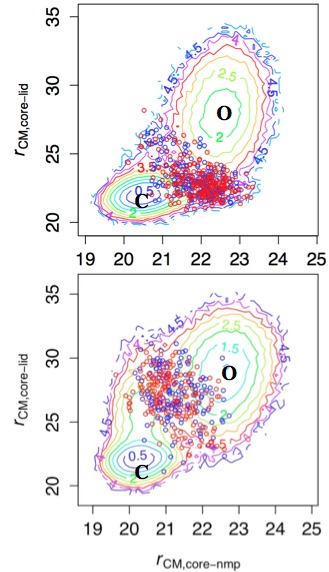
TS ensemble and free energy landscape in Adenylate Kinase:
Coarse-grained (Double-well Go model) simulations suggest that the dominant pathway for the Open/Close transition changes upon ligand binding (top vs. bottom). This is rationalized by the fact that the interface between the CORE and LID domains features many ligand-mediated interactions. Therefore, in the absence of ligand, the highly flexible LID domain closes at the late stage of the transition due to entropic reason. In the presence of ligand, however, the LID domain closes first to form many enthalpically favorable interactions with the CORE domain. The enthalpy/entropy compensation phenomena is expected to be prevalent in allosteric transitions. See, Daily et al., J. Mol. Biol. 400, 618-631 (2010)
A coarse-grained model for water with correct electrostatics:
Development of coarse-grained (CG) models has become increasingly important in the study of complex processes such as lipid self-assembly. While popular models such as MARTINI work well for many membrane problems, it remains challenging to develop CG models that treat processes in which electrostatics play an important role, such as binding of highly charged peptides to lipid membrane. We have filled this important void by the development of a Big Multipole Water (BMW) model. See, Wu et al., J. Phys. Chem. B , 114, 10524-10529 (2010)
An effective implicit solvent model for rxns involving highly charged species:
Although implicit solvent models based on Poisson(-Boltzmann) have been used in quantum chemistry for many years, it remains challenging to describe chemical reactions in solution that involve highly charged species. This is an important challenge to tackle because many biologically relevant reactions, such as phosphoryl transfers, implicate highly charged groups. To this end, we have adopted a simple but effective scheme in which the atomic radii are taken to be linearly correlated to the atomic charges of the QM solute. Implementation of this scheme in the context of SCC-DFTB led to encouraging results. See, Hou et al., J. Chem. Theo. Comp. , 6, 2303-2314 (2010)
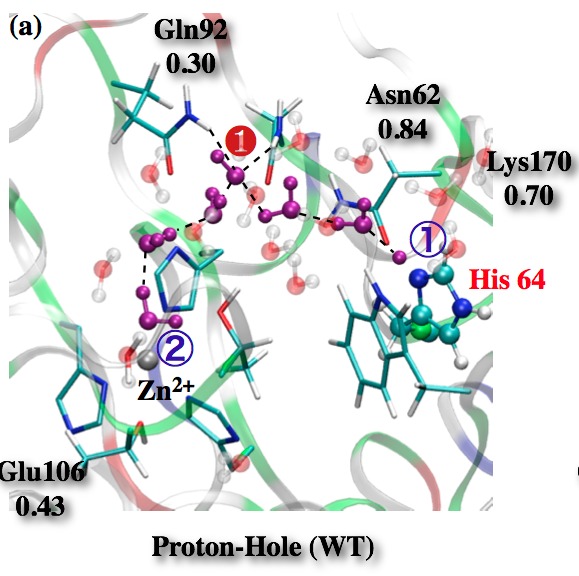
Proton hole pathway for proton transfer in Carbonic Anhydrase II (CAII):
QM/MM simulations point to the possible involvement of a “proton hole pathway” for proton transfer in CAII, which is microscopically distinct from the commonly discussed Grotthuss pathway for long-range proton transfers in solution and biomolecules. Although the relative weights of the proton hole and Grotthuss pathways remain difficult to characterize using current computational methods, QM/MM calculations suggest that Phi-like analysis might be used to probe the importance of the proton hole pathway in experiments. See, for a review, Riccardi et al., BBA-Prot. & Proteomics, 1804, 342-351 (2010)
Quantitative computation of small solute effects:
Comparison of preferential interaction coefficients (Gamma_23) for urea and glycine betaine around small peptides from molecular simulations and thermodynamic measurements suggests that popular molecular force field (e.g., CHARMM) is qualitatively reliable for describing the interaction between small solutes and biomolecules, relative to their interactions with water molecules. For quantitative agreement, however, refinements of force field parameters are necessary. The computations also support an additive thermodynamic model based on surface area. See, Ma et al., Biochemistry , 49, 1954-1962 (2010)
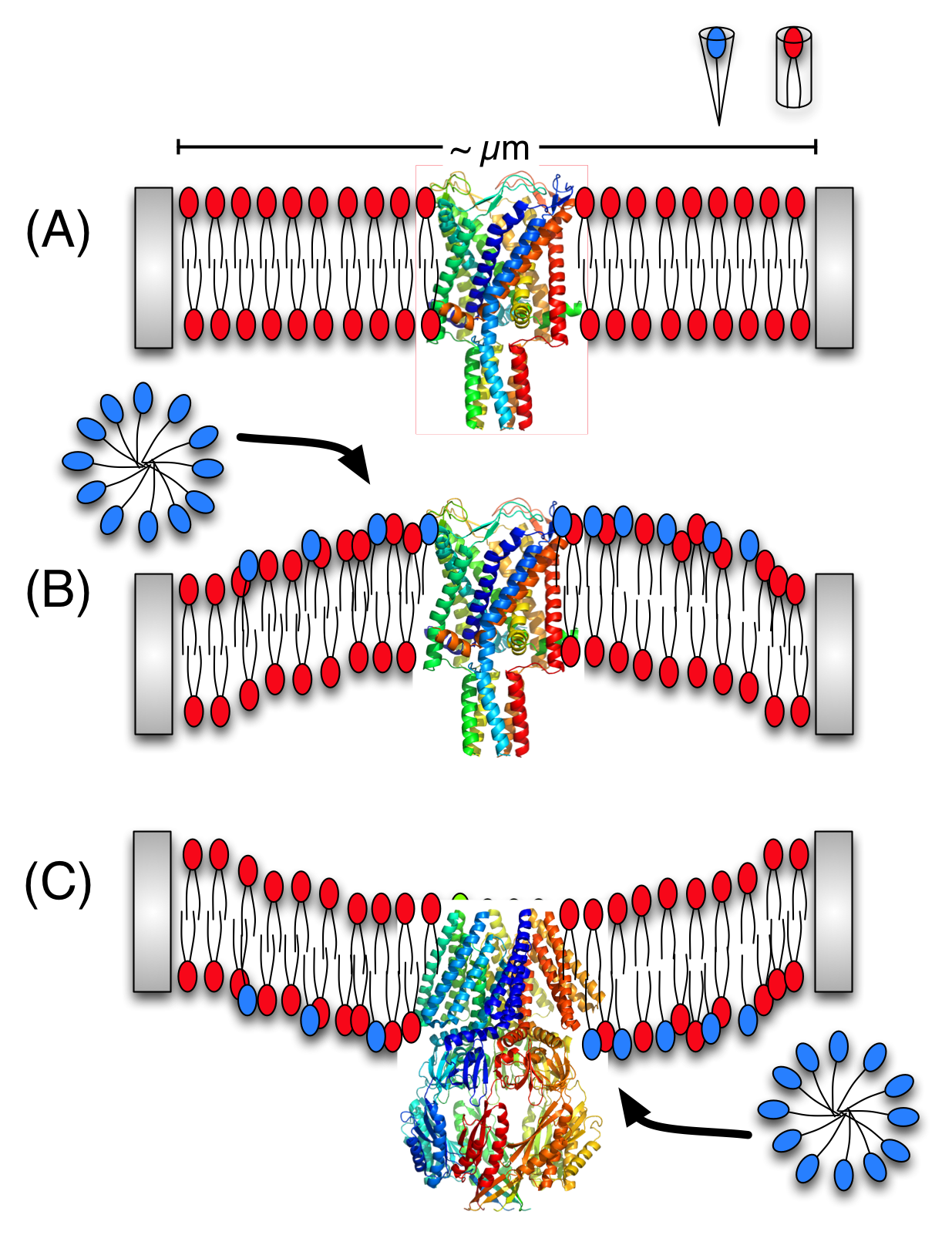
Activation of mechano-sensitive channels by asymmetric LPC incorporation:
Although it is well-known that mechano-sensitive channels can be activated by not only membrane tension but also asymmetric incorporation of cone-shaped lipids into memembrane patches, the underlying mechanism is not clear. Using a coarse-grained lipid model (MARTINI), we developed a simple scheme to generate curvature in membrane mixtures and computed the resulting local pressure profiles. The analysis indicates that perturbation in local pressure profile doesn’t depend on the local curvature per se, but on the local lipid packing (area-per-lipid). For more details, see, Yoo et al., Biophys. J. 97, 2267-2276 (2009)
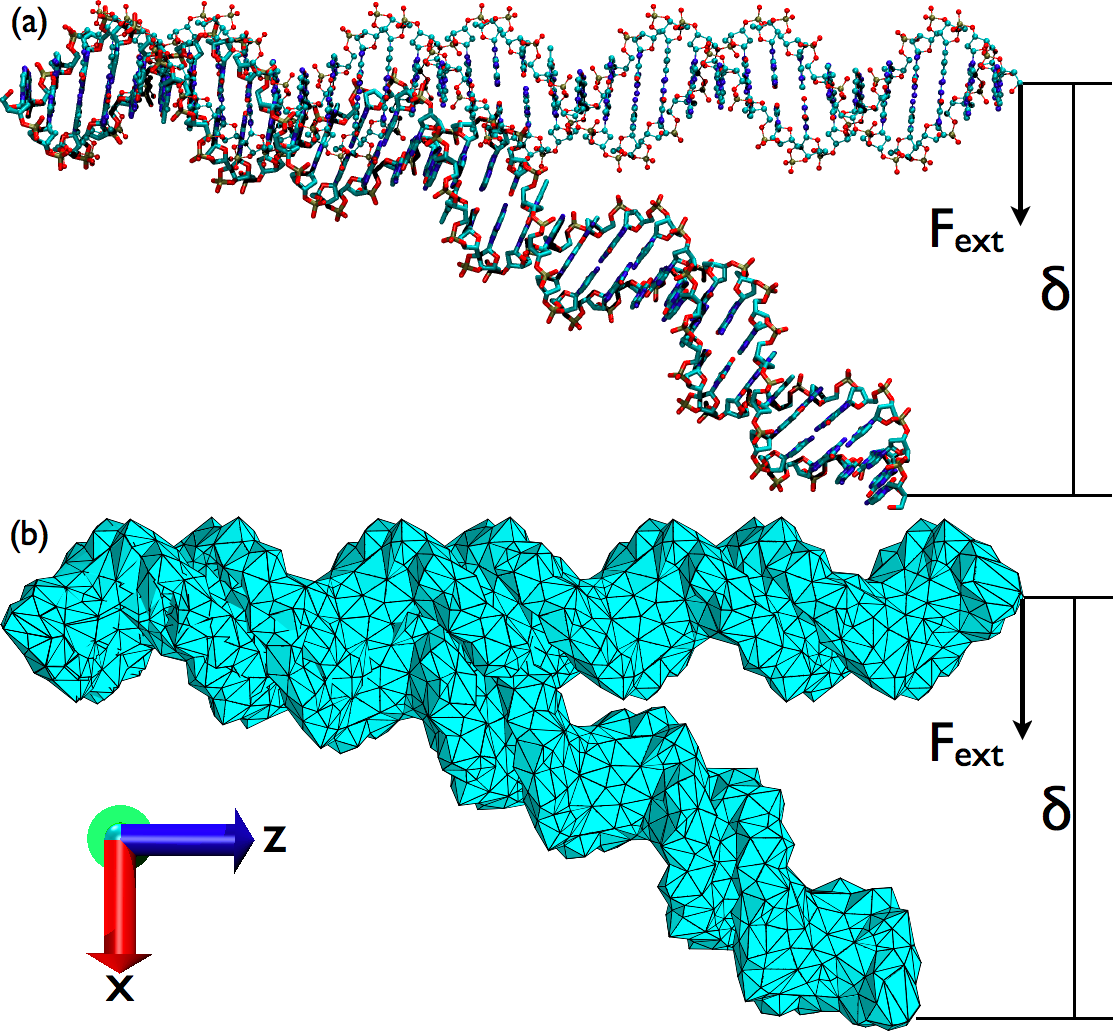
Combined continuum mechanics/continuum electrostatic (CM/CE) models for macromolecular deformation:
combined CM/CE calculations using a simple Debye-Huckel model reproduced the salt-dependence of DNA bendability. With the more sophisticated Poisson-Boltzmann model, however, numerical issues were observed for systems with a high aspect ratio (e.g.,a very long DNA). Therefore, although the combined CM/CE model is promising for studying mechanical deformation of macromolecular complexes, care is needed to ensure numerical stability. See, Ma et al., Biophys. J. 96, 3543-3554 (2009)
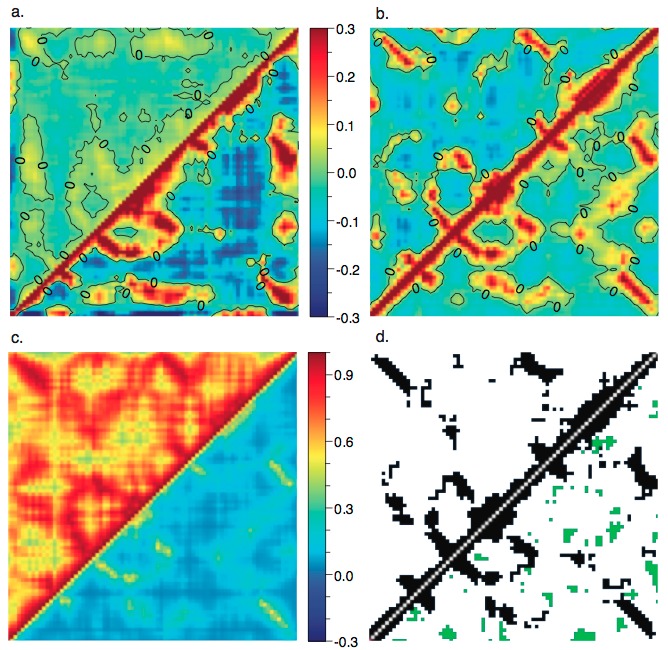
Atomic correlation from different elastic network models with different boundary conditions:
Atomic correlations are sensitive to the treatment of (a) cutoff and (b) crystal contacts; including the lattice vibrations (through Born-von Karman boundary condition) has an important impact on the calculated atomic correlations. For more details, see, Riccardi et al., Biophys. J. 96, 464-475 (2009)
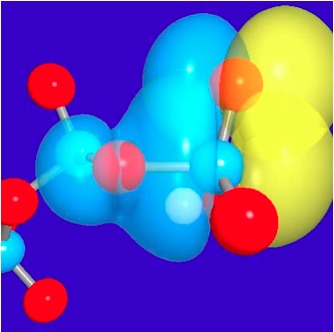
Illustrations of the anomeric effects in tri-phosphate:
3D representations of the pre-orthogonal natural bond orbitals (PNBO) for the lone pair of a nonbridging oxygen of the gamma-phosphate and the scissile P-O anti-bonding orbital in ATP. Red spheres indicate oxygen atoms and blue spheres indicate phosphorus atoms. See, Yang et al., J. Phys. Chem. A (Pitzer issue) , 113, 12439-12446 (2009)
Proton Release Group (PRG) in bacteriorhodopsin (bR):
QM/MM simulations suggest that the PRG is NOT a protonated water cluster, but rather a pair of conserved Glutamate residues in that region, between which the stored proton is delocalized; the delocalization gives rise to the unusual continuum band in the IR spectra. See, Phatak et al., PNAS, 105, 19672-19677 (2008)
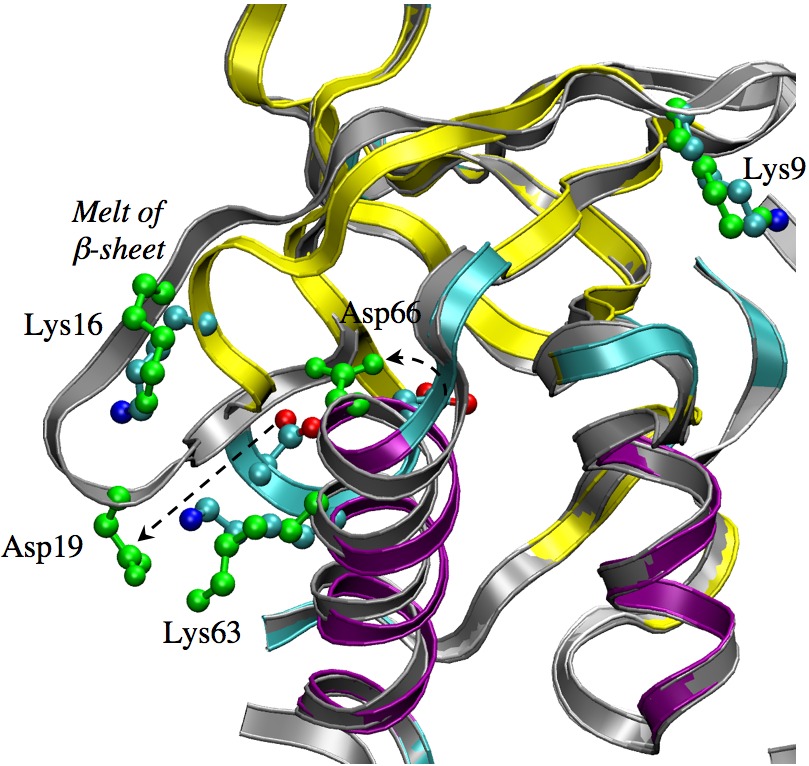
Structural responses observed for the titration of Asp66 in SNase mutant:
(grey/green vs. color-coded x-ray structure). Microscopic mechanism for the titration of buried protein residues is challenging to reveal because the titration is likely coupled to significant structural transitions; neither the magnitude or location of the transition is straightforward to predict, making molecular simulations uniquely powerful in this context. However, enhanced sampling technique is likely essential for capturing the correct structural transitions, an issue highly relevant to the understanding of proton transport systems and pH gated ion channels. See, Ghosh et al., J. Phys. Chem. B 112, 8387-8397 (2008)
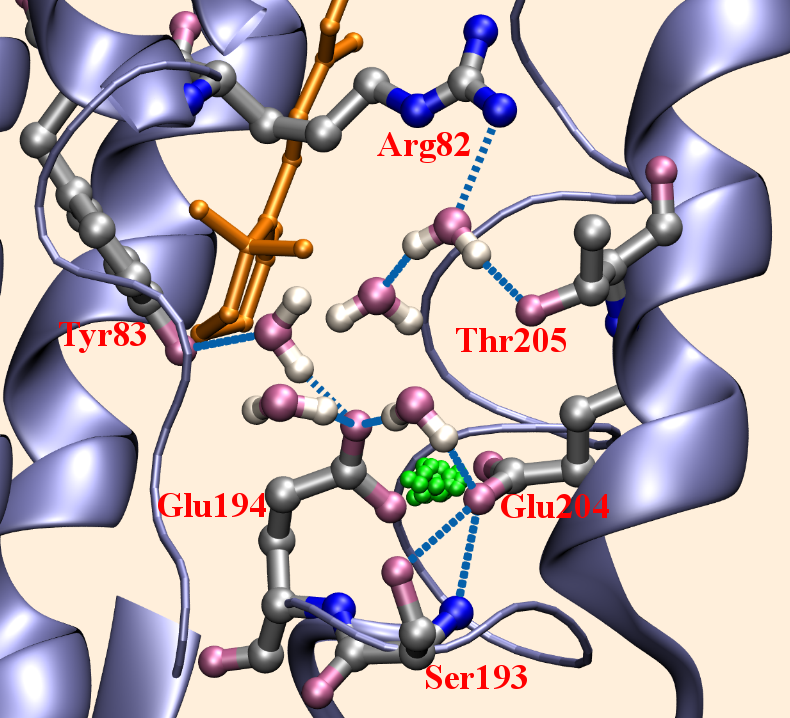
Activation of ATPase in myosin requires extensive structural changes:
By combining QM/MM and classical PMF simulations, we demonstrated that the transition from the post-rigor state to a structurally stable closed active site (which apparently is critical to efficient ATP hydrolysis) relies on not only the displacement of SwII but also more extensive structural rearrangements in the nearby region. In other words, structural transitions remote from the active site can play an active role in regulating the hydrolysis of ATP, rather than passively responding to structural changes in the active site; this is likely a general feature shared among biomolecules whose function relies on mechanochemical coupling between distant sites. See, Yang et al., J. Mol. Biol. 381, 1407-1420 (2007)
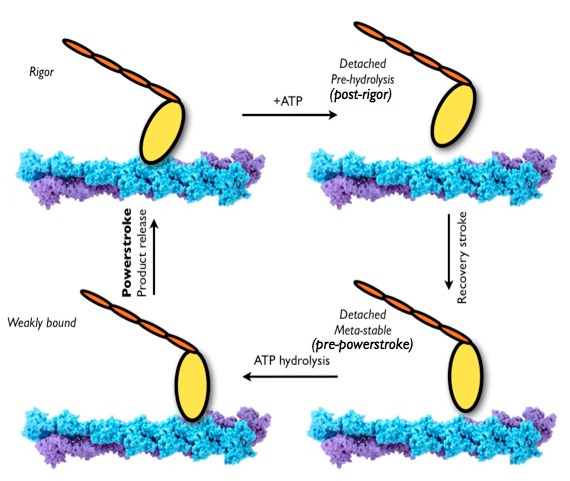
The mechanism of recovery stroke in the myosin motor domain:
The hydrolysis of Adenosine triphosphate (ATP) provides the energy for most life processes, including the motility of molecular motors. How the chemical energy of hydrolysis is converted into mechanical work in these fascinating “nanomachines” is a central question that has only been answered in an outline form for almost all molecular motors. The fundamental challenge is that the working cycle of molecular motors involves processes of different physicochemical natures and scales, including ATP chemistry and protein structural transitions of diverse magnitudes. Combined with previous computational studies from this lab, molecular dynamics simulations help identify energetic and structural properties of myosin, a prototypical molecular motor, that are essential to its energy conversion function. See, Yu et al.,PLos Comp. Biol.3, 199; 214 (2007)
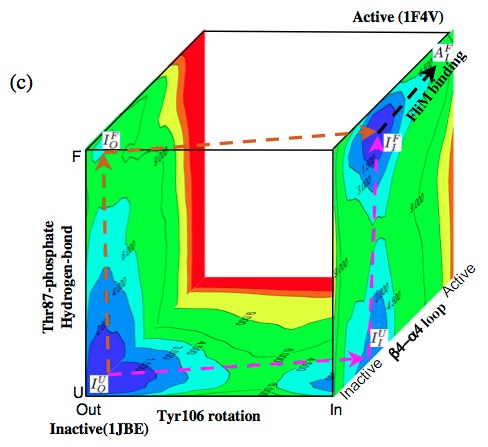
Mapping the free energy landscape of CheY activation:
A three-dimensional scheme that illustrates the energetics and possible pathways for CheY activation based on the computed 2-dimensional PMFs along the key degrees of freedom. The results show that structural transition at the response site (Tyr 106 isomerization) can occur prior to the so-called activation event (Thr 87-phosphate hydrogen-bond formation). This suggests that the Tyr orientations are in equilibrium and that the active conformation is stabilized by Thr 87-phosphate hydrogen bond formations; kinetically, either event can occur first. In other words, these studies suggest that the mechanism of CheY activation has features of both the MWC and Pauling-KNF descriptions. See the recent review of Cui and Karplus, Protein Science, 17, 1295-1307 (2008) and the original work below.
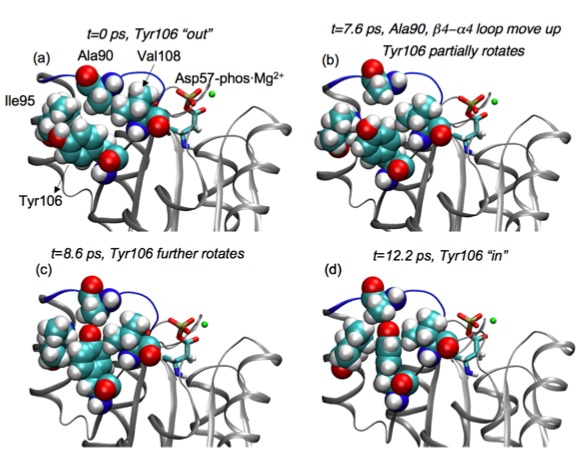
Unbiased simulation of conformational transition in a signaling protein:
Transition path sampling (TPS) is used to harvest unbiased activation trajectories of a small signaling protein, CheY. The results are in direct contrast to those from a previous biased molecular dynamics simulation, highlighting the value of TPS in the study of complex transition mechanism in proteins. See, Ma et al., J. Am. Chem. Soc.129, 10261-10268 (2007)