Research
Active Projects
Professor David Coker and Professor John Porco
-
ParaTemp
Building upon the work of Dr. Thomas Heavey and in collaboration with the Coker research group and Porco research group, I am developing an “organic chemist’s” interface to sophisticated theoretical techniques used to sample the conformational space of complex molecules; in particular replica exchange molecular dynamics, otherwise known as parallel tempering.
Professor Linda Doerrer
-
The Electronic Structure of Square-Planar Co(III) Systems
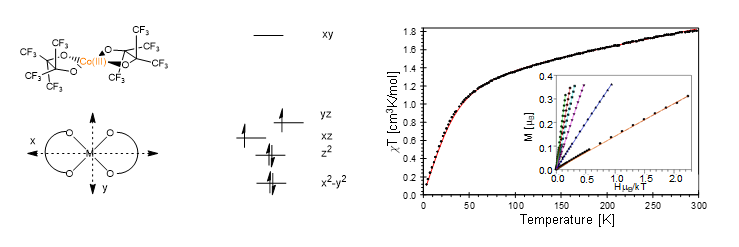
Spurred on by the interesting electronic structure that was observed for the square planar Co(III)(pinF)2 system that was reported by the Doerrer group (10.1039/C8CC04464C), I am currently doing a comprehensive investigation into the electronic structure of Co(III) square planar systems. In particular, I am investigating the detailed structural features that affect the relevant energy levels that result in the significant spin-orbit coupling observed in these systems. I am also looking at the electronic feature that causes the unexpected rhombicity observed in some of these systems.
Professor Aaron Beeler
-
Development of Scripts For QM Descriptor Production
PhD candidate Doug Fraser is involved in an ambitious multi-component project. This project will develop a high-yield instrumental technique that rapidly identifies suitable reactants that will be combined with a database of structural predictors augmented by quantum-mechanical descriptors that will then be mined by the statistical department at BU to identify key descriptors that help predict reactivity. I am assisting Doug in developing scripting tools that will allow high-throughput calculations that automatically organizes and extracts descriptors so that the data can be mined by the statisticians.
Professor Mark Grinstaff
-
FMO Theory For Protecting Group Variation in Glycal+TCAI Production of beta-Lactams.
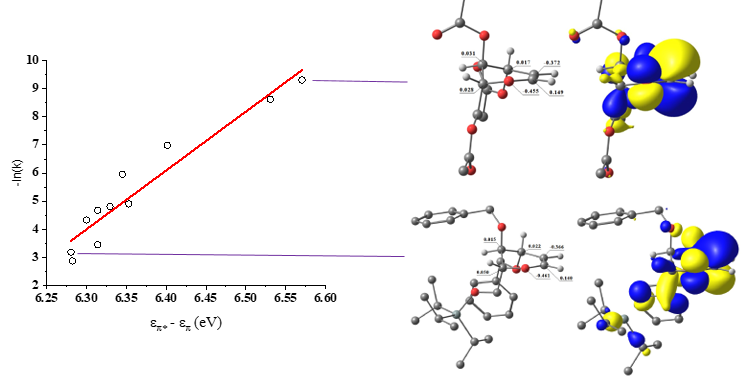
PhD candidate Anant Balijepalli made the important observation that the [2+2] cycloaddition of glycals with TCAI is correlated with the electron-withdrawing power of glycal protecting groups. Anu synthesized a library of 18 electron-rich alkenes (glycals) with varied protecting groups to systematically elucidate the factors that influence their reactivity towards an electron-poor isocyanate (trichloroacetyl isocyanate). The experimentally determined reaction rates exponentially correlate with the computationally-determined HOMO-LUMO gap and NBO (natural bond orbital) valence energies. The electron withdrawing ability of the protecting groups, but not bulk, impact the electron density of the glycal allyloxocarbenium system when oriented pseudo-axially (i.e., stereoelectronics). In this conformation, ring σ* (C-O) orbitals oriented anti-periplanar to the allyloxocarbenium system decrease glycal reactivity via negative hyperconjugation as protecting group electron withdrawal increases. Transition state calculations reveal that protecting group stereoelectronics direct the reaction to proceed via a concerted, asynchronous mechanism through a zwitterionic species, supported by a rate acceleration in polar reaction solvents (e.g., acetonitrile and nitromethane).
Professor John Straub and Professor Mark Grinstaff
-
Mapping the PES of Poly-Amido-Saccharides.
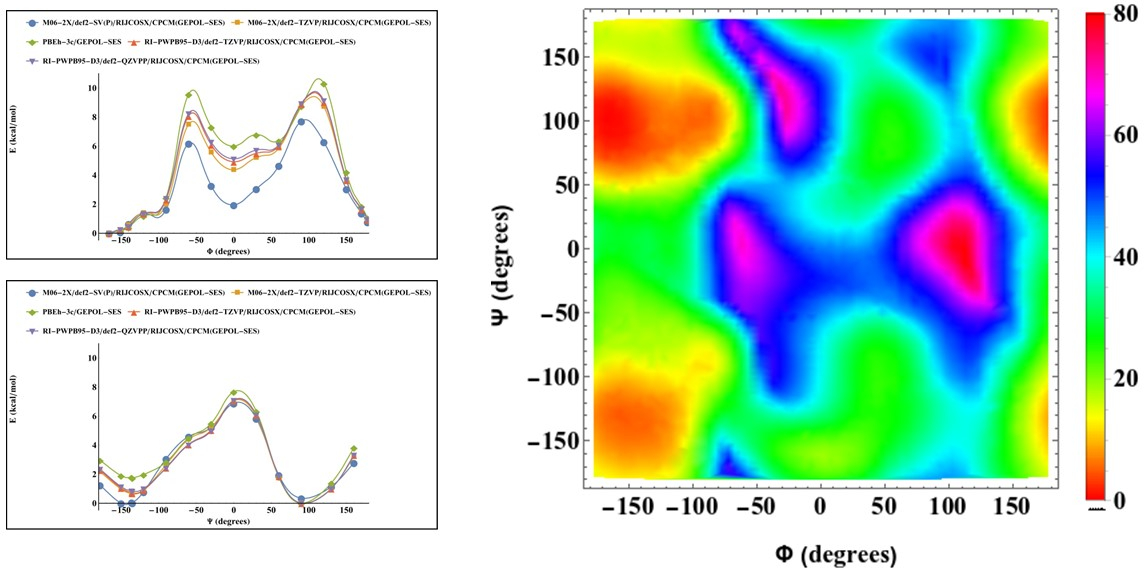
Hannah Caputo of the Grinstaff research group and PhD candidate Asange Bandara of the Straub research group collaborated on a project to elucidate the conformational space of sulfated polysaccharides and form a predictive model for the reactivity of the monomers and three-dimensional conformations of the polysaccharides. While Hannah has moved on, Asanga is continuing the research and has benchmarked and verified the force field in his work against QM calculations performed by Hannah and myself.
Long-Term Projects
-
EPR Spectral Simulation
-
ORCA NBO
-
Customizable Interfaces For Computational Engines
-
Direct Dynamics and Variational TST For ORCA
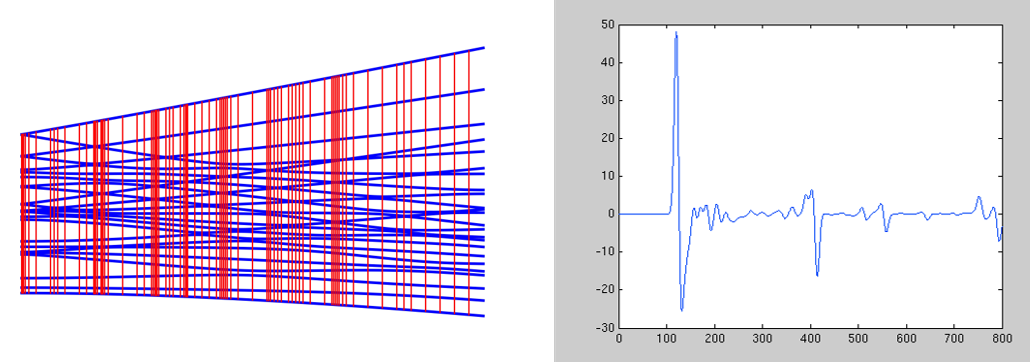
Based on work done in my dissertation, I am continuing to develop a software package written in C++ that will do a full diagonalization of the Spin Hamiltonian matrix with a user-friendly interface. This program will be available to all groups in the department upon completion and includes helpful ‘guides’ for students. The simulations can also be combined with ESS software to automate the dual experimental fitting/theoretical prediction of EPR spectra. This program is an enhancement on currently available software packages in that it is 1) parallelized; 2) highly efficient; 3) able to run in a “black-box” fashion.
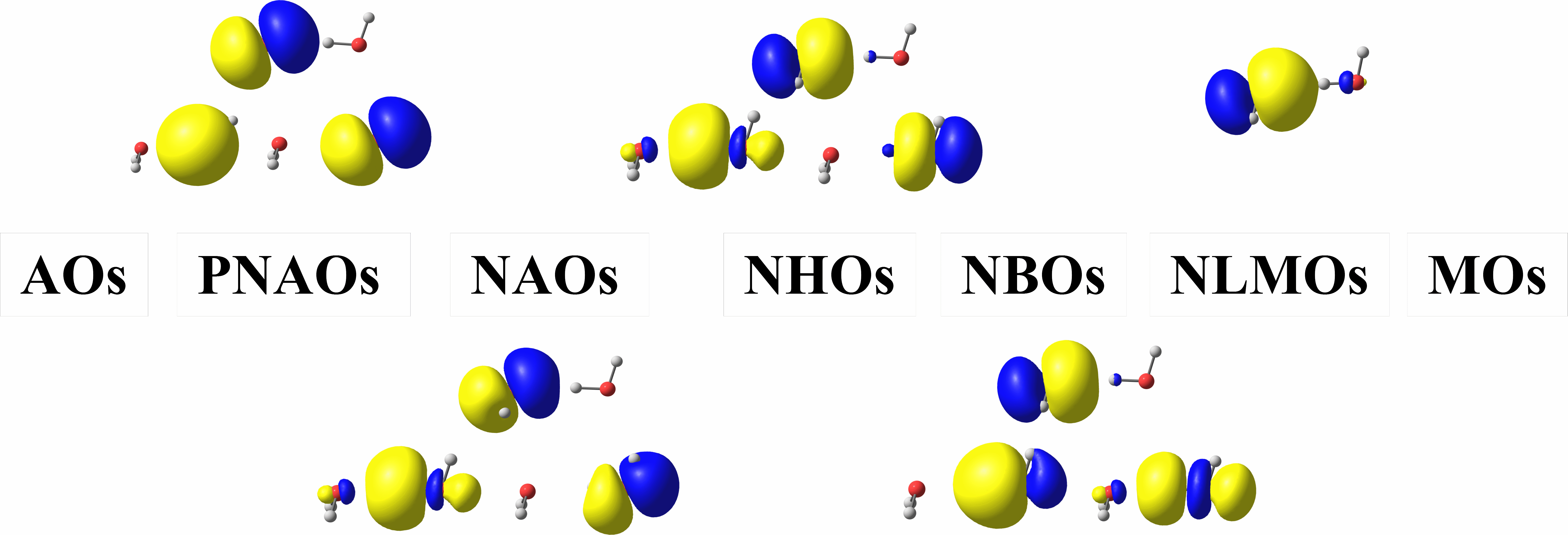
Although the latest version of the ORCA ESS contain NBO options when linked to a exiting NBO executable, the ability of ORCA to interface with NBO is rather limited, and it requires a separate purchase of the GENNBO code. I am writing the NBO code from scratch with C++ and the Eigen library to give full control of all NBO options to users who prefer the ORCA ESS. In addition to giving greater access to the NBO functionalities and spurring further modifications and analysis tools based on NBO localization constructs, the new program will be able to interactively cooperate with ORCA to perform functions such as $DEL and $NEDA. I currently have a functional subroutine that will generate NBO formatted archive files from an ORCA output file that can be used as input for the standalone GENNBO executable. Please contact me if you’d like to use the program.
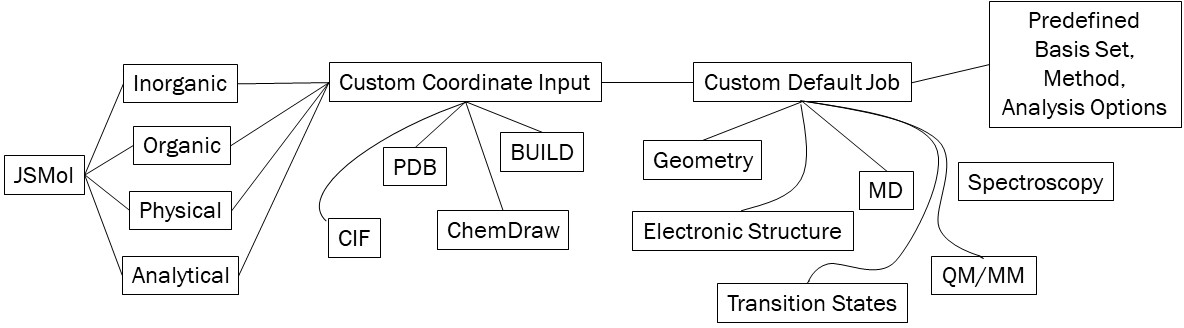
Although they are sometimes treated as such, computational methods are not “black-box” methods. Particularly in light of the density functional stew that is available, researches must robustly identify the proper computational technique to approach different problems. The goal of this project is to create tailored “black-boxes”. Based on modern literature and my expertise, interfaces for different research will be designed so that the desired outcome is robustly achieved. Each research group generally studies systems that can be broadly grouped together, and thus each group will contain their own interface that reduces the bottleneck of incoming students mastering the command line and basic quantum chemistry before obtaining results.
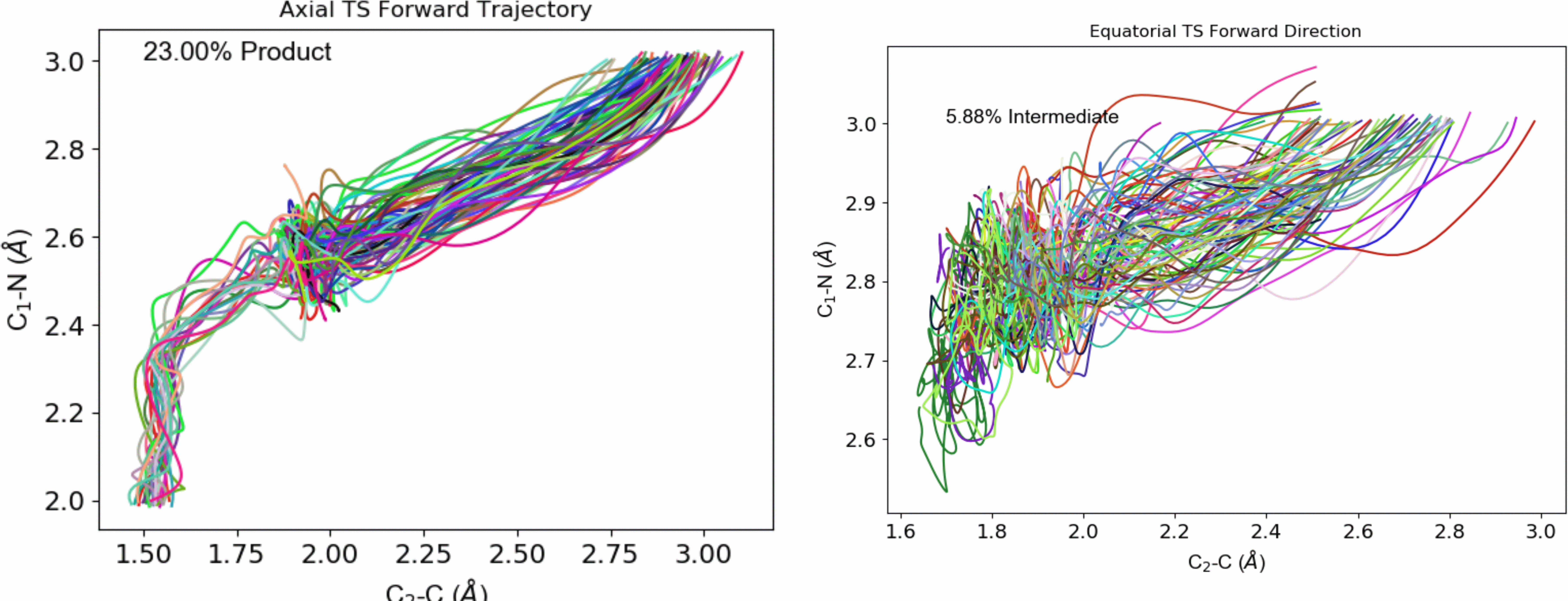
I have completed a beta version of a C++ program that interfaces with the ORCA ESS to perform quasi-classical direct dynamics calculations. The program is largely based on Singleton’s PROGDYN, but is being extended to include Variational TST calculations based on the work of Truhlar et al. While ORCA recently developed their own MD capabilities that are built into the binaries, my work gives users full access to the dynamics options, and it also can be tailored for specific systems. Shown above is a representative results displaying a system that is predicted by classical TST to preferrably pass through a transition state that is not the experimentally determined reaction pathway. Please contact me if you’d like to obtain the program or would like a custom modification made.
Other Research Interests
Electronic Structure of Non-Innocent Ligands

The pyridine-2,6-diimine (PDI) ligand has a wealth of documentation pertaining to its uncanny ability to stabilize a number of transition metal reactions. This interest was largely prompted by the works of Gibson and Brookhart on cobalt and iron-based olefin polymerization using PDI-catalyzed systems. Following these studies, other groups entered the foray and immense progress has been made.
Several groups have further explored the ability of PDI systems to catalyze olefin polymerization/oligomerization, dimerization, and hydrogenation. Chirik et al. and others have shown that PDI systems promote a number of other functionalizations including hydroboration reactions and hydrosilations. There have also been a large number of studies demonstrating the ability of PDI systems to activate C-H bonds and catalyze C-C bond formation. Burger et al. demonstrated the ability of a PDI-anchored Iridium system to catalytically promote nitrogen fixation. Still other groups have shown N-N bond cleavage promoted by PDI systems , C-O functionalization, O2 activation, and N-H activation. Gilbertson recently demonstrated the ability of the Fe2+(PDI)X2 (X = Cl-, Br-) system to catalytically reduce carbon dioxide to carbon monoxide, a highly desirable reaction.
Much of the work mentioned above used Density Functional Theory (DFT) or a combination of DFT and Broken Symmetry (DFT-BS) to theoretically characterize the PDI systems. There have been other successful applications of DFT-BS techniques to PDI systems that were not mentioned above. There are a surprisingly small amount of multi-reference investigations into the electronic structure of the PDI ligand and transition metal complexes containing the PDI ligand. Guihery et al. used CASSCF(2,2) and difference dedicated configuration interaction (DDCI) techniques to study how the electronic structure of PDI was affected by different point charges located at the metallic position. Burger et al. used MRMP2(10,10) technique to characterize an Iridium-Nitride triple bond stabilized by the PDI scaffold. This complex is involved in catalytic N2 fixation.
I have shown the following: First, while DFT-BS results are useful and often correlate well with experimental results, they can also be ambiguous. Different DFT-BS approaches will be energetically similar, but only one of the approaches agrees with experiment. This is a serious problem if one is studying a system that has not been characterized experimentally. Second, the choice of active space when studying PDI-Fe(II) systems must be made very carefully. If the proper choice of active space is not made, the electronic structure obtained via MRMP2/CASSCF methods does not correlate with experiment. Third, I have shown the correct electronic structure of Fe2+(PDI)X2 (X = Cl-, Br-) based on MRMP2(14,14) results.
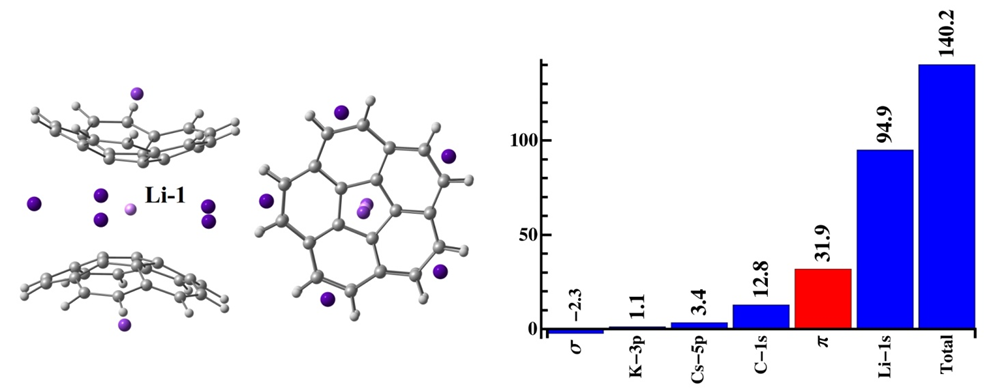
Electronic Structure of Curved Polyaromatic Systems