New paper – Light Chain Stabilization: A Therapeutic Approach to Ameliorate AL Amyloidosis
Morgan, Buxbaum and Kelly; Hemato 2021, 2(4), 645-659
A major goal of our research is to create small molecule drugs to stabilize antibody light chains and suppress their misfolding and aggregation, as a potential therapy for light chain (AL) amyloidosis. Along with my collaborators, Professors Jeff Kelly and Joel Buxbaum at Scripps Research, I wrote a review article about our aims for the project, why we think that it could benefit patients, and our progress to date (open access).
Jeff, who was my postdoctoral advisor at Scripps, recently won the Breakthrough Prize for showing that unstable proteins can cause amyloidosis and that drugs that stabilize these proteins can benefit patients. The outcome of these studies was Pfizer’s drug tafamidis, which is used to treat ATTR amyloidosis, where aggregation of unstable transthyretin (TTR) protein leads to heart and nerve damage. To hear more about the discovery of tafamidis, check out the Scripps Front Row lecture that Jeff gave recently.
It’s worth taking a moment to ask why tafamidis might be considered more significant than the mRNA-directed gene silencing drugs, patisiran and inotersen, which are also approved for treatment of ATTR amyloidosis. To me, the key distinction is that RNA silencing was a therapeutic mechanism looking for a disease, which can be applied to many potential targets. They represent incredible progress, but the strategies were not designed with amyloidosis in mind. TTR has been the target of choice because it’s made in the organ that can be most reliably targeted with the silencers, which is the liver. Whereas tafamidis was designed as a stabilizer of TTR to test a specific therapeutic mechanism with broader implications, that pharmacological stabilization of otherwise unstable proteins could correct genetic disease. One of the components of the cystic fibrosis therapy, trikafta, works in the same way, and there is potential for other disease-associated proteins. So based on this clinical utility of tafamidis for ATTR amyloidosis, we aim to create analogous drugs for AL amyloidosis.
AL amyloidosis treatments are designed to eradicate the clonal plasma cells that secrete amyloidogenic light chains. New therapies do this very well, but many patients continue to experience symptoms from amyloid deposits. For example, only half of patients treated with daratumumab had an “organ response” (improvement in their heart or kidney function) after six months of therapy Part of the reason for this is that amyloid deposits are not cleared by the body, so medicines that aim to remove amyloid fibrils are urgently needed. But this is difficult and clinical data so far is, at best, encouraging rather than spectacular.
Another possible factor is that even low levels of light chain production by surviving clonal plasma cells can continue to drive amyloid formation. We know that more effective eradication of these cells leads to better outcomes, and that long-term remission is possible. So if we can prevent aggregation of whatever light chains remain, either before or after cytotoxic therapy, this should reduce growth of amyloid deposits and allow damaged tissues to recover. We see stabilizers as a complement to existing therapies – stopping production of clonal light chains would still be the priority.
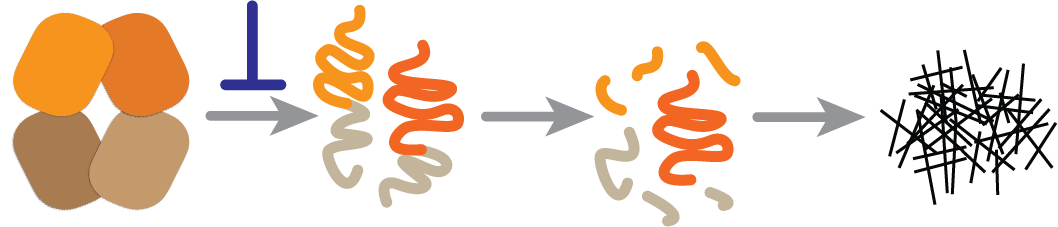
A key conceptual advantage of stabilization is that it acts early in the disease process (the blue inhibition symbol in the figure). We don’t fully understand all of the ways that light chains cause tissue damage, whether as amyloid fibrils, misfolded oligomers or non-native monomers or dimers. But stabilizing the native, folded light chain should prevent formation of all non-native toxic conformations.
We have made some important progress towards drug candidates, but there is still a long way to go. A big milestone is demonstrating that small molecules can bind to light chains and protect them from proteolysis with drug-like, nanomolar affinity. When we set out, we didn’t know if would be possible at all.
Importantly though, there are a number of questions that we can’t answer without clinical trials, so we need to be cautious about how we approach treating patients. There are no animal models that recapitulate all of the features of systemic amyloidosis, so we can’t look for efficacy in mice. We need to make sure that our molecules are as effective as we can make them in lab assays – ensuring that they bind tightly and specifically to light chains, and that they are non-toxic. But we will need a clinical trial in humans to learn whether the molecules are effective enough, and whether stabilizing light chains actually suppresses amyloid formation.
We are confident that we can make a molecule that is safe for people, and stabilizes light chains. And we are cautiously optimistic that stabilization will suppress amyloid formation and be of benefit to patients. Let’s see how far we can take this idea.