Top Ranked ENG Algorithms Could Reveal New Cancer Drug Target
by Mark Dwortzan
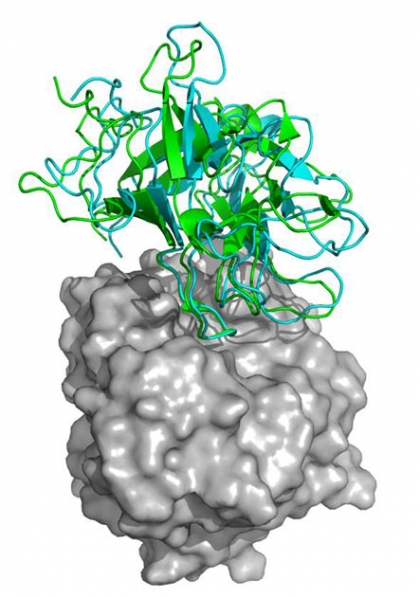
An interdisciplinary College of Engineering team has developed computational methods to predict the structures that form when two cellular proteins interact. Judged the world�s best at a major international competition, these methods have drawn more than $3 million in NIH funding.
An interdisciplinary College of Engineering team has developed computational methods to predict the structures that form when two cellular proteins interact. Judged the world’s best at a major international competition, these methods have drawn more than $3 million in NIH funding.
An interdisciplinary team of College of Engineering faculty members—Professor Sandor Vajda and Research Assistant Professor Dima Kozakov (both BME), Professor Ioannis Paschalidis (ECE) and Associate Professor Pirooz Vakili (ME)—has developed a family of powerful optimization algorithms for predicting the structures of complexes that form when two cell proteins bond together—structures that, in some cases, generate erroneous cell signaling pathways that can trigger cancer and other inflammatory diseases. Recognizing the promise of these computational methods to advance new approaches to combating these illnesses, the National Institutes of Health (NIH)-National Institute of General Medical Sciences (NIGMS) recently approved a $1.6 million, five-year grant that will enable the researchers to continue refining them.
A joint effort of Boston University’s Center for Information and Systems Engineering and Biomolecular Engineering Research Center, the project combines Paschalidis’ and Vakili’s expertise in optimization and systems theory with Vajda and Kozakov’s knowledge of biophysics and bioinformatics.
“Given the three-dimensional structure of two proteins, you’d like to predict with great accuracy the structure of the complex formed once these two proteins bind,” said Paschalidis, who compares the process to characterizing all the possible structures that pairs of Lego blocks can form out of an initial set of 1,000 blocks. “Based on laws of thermodynamics, we’ve developed optimization algorithms that have succeeded in doing just that.”
Applying those high-precision algorithms to a related effort, Vajda and Kozakov are also collaborating with BU Associate Professor Adrian Whitty, Professor John Porco, Professor Karen Allen, and Research Assistant Professor Aaron Beeler (all Department of Chemistry); and Professor Gilmore (Department of Biology) to identify small molecules—potential drugs—that can disrupt protein-protein interactions (PPIs) that produce structures that may provoke illness.
Operating under a separate, four-year, $1.6 million NIH-NIGMS grant, the BU team aims to develop synthetic molecules that inhibit PPI targets at sites identified by Vajda’s and Kozakov’s innovative computational approaches—without introducing any harmful side effects. In particular, the researchers are attempting to design novel molecules that can block chronically hyperactive cell signaling pathways found in human inflammatory diseases and cancers.
Despite decades of effort by the pharmaceutical industry, it has proven extraordinarily difficult to develop oral drugs that inhibit PPIs, Vajda observed. “Current protein-based drugs can disrupt PPIs, but only on the surface of cells,” he said. “But 90 percent of PPIs occur within the cell, and these are the interactions that we’re working to disrupt.”
ENG Algorithms Judged the World’s Best
The NIH is funding the development and application of these advanced computational methods because scientists cannot easily observe the complexes formed by protein-protein interactions. Many biologically important PPIs produce fragile complexes that do not remain intact long enough to be subject to direct experimental analysis, but optimization algorithms such as those developed by the ENG team can determine the structure of these complexes with great accuracy based on the structures of the component proteins.
As the team improves its methods, they will ultimately be incorporated into Vajda’s and Kozakov’s protein-protein docking server ClusPro, a website to which any user can submit the three-dimensional coordinates of two proteins and receive a supercomputer-calculated prediction of the structure of the complex formed by those proteins. ClusPro has been used by over 3000 research groups worldwide, and generated over 100 structures reported in the scientific literature.
In a worldwide blind prediction experiment conducted by CAPRI (Critical Assessment of Predicted Interactions) in Barcelona last December, judges determined that the ENG team’s computational methods delivered the best performance in modeling selected protein-protein complexes when compared against 64 other predictor groups that combine software models with human analysis, and that ClusPro also performed better than nine other competing automated docking servers, which rely on software alone. Results of the CAPRI experiment will be published in a special issue of the journal Proteins: Structure, Function, and Bioinformatics in November.